ABSTRACT
Malaria is one of the major causes of morbidity and mortality in tropical and sub-tropical parts of the world. Plasmodium falciparum polyadenylate binding protein (PABP) plays a vital role in the stable RNA accumulation of host cells during P. falciparum malaria parasite infection. This protein mediates the liver stage invasion of the parasite by binding to poly-A tail of the mRNA using its globular domains that contain RNA-recognition motifs (RRMs) which regulate mRNA stability and protein translation. An in-silico analysis and modeling of the P. falciparum PABP was carried out to elucidate the physicochemical properties, disease-causing regions, the protein-protein interactions as well as the predicted structure of the protein. The primary and secondary structural features of the protein were calculated by ProtParam and SOPMA, respectively, which revealed the protein is composed of random coils (41.71%), a-helix (36.23%), extended strand (14.06%), and b turn (8.00%). The three-dimensional structure of P. falciparum PABP was not available as yet at PDB. Therefore, homology models for these proteins were developed using SWISS-MODEL, PHYRE2, and I-TASSER Web Server. The models were visualized with RASMOL and validated using PROCHECK, Verify3D, and QMEAN for reliability. 92.64% of the residues in the predicted model have averaged 3D-1D score ≥ 0.2 which indicates that the predicted model is compatible with the sequence. Protein-protein and residue-residue interaction networks were generated by the STRING and RING servers, respectively. 3DLigand server was used to analyze binding sites of the modeled PABP. This predicted structure of P. falciparum PABP will make an important contribution towards better understanding of the functions of the protein in translation regulation in the parasite and may also provide targets for novel therapeutic candidates.
Key words: Malaria, poly A-binding protein, bioinformatics analysis, Plasmodium falciparum, PfPABP.
Malaria is an infectious disease that is highly distributed in the tropical and subtropical regions of the world, and it is caused by the parasite of the genus Plasmodium. Malaria is one of the leading causes of death by a communicable infectious agent, claiming 405,000 lives globally in 2018 with 67% of the total mortality being children under 5 years (WHO, 2019). The malaria parasite has a complex life cycle, involving an invertebrate vector (mosquito) and a vertebrate host. Each stage of the parasite’s life cycle involves arrays of genes whose expressions are tightly regulated.
The regulation of gene expression and the synthesis of proteins is pertinent for the parasite to carry out its sophisticated developmental program since the parasite does not know when it would be transmitted from the mosquito to the human host and vice versa (Coulson et al., 2004; Cui et al., 2002). The genome of the parasite is deficient in transcriptional regulators (Coulson et al., 2004), which implies that post-transcriptional regulation are important in regulating the expression of the parasite’s genes as well as in protein synthesis (Reddy et al., 2015). Gene regulation via a post-transcriptional mechanism is dependent on RNA-binding proteins, which play important roles in the parasite’s biology especially during the transmission stage (Bunnik et al., 2016). The transmission stages (sporozoites and gametocytes) must remain inactive, stable, and infectious in the vector or host for an extended period before a mosquito may bite and pick them up again (Minns et al., 2018). At these stages, mRNA which is important for development in the host or vectors are kept translationally repressed (Reddy et al., 2015)so that rapid protein synthesis can occur presumably when the opportunity to infect arises (Cui et al., 2015). These translationally repressed transcripts are stored in punctate storage granules within the cytoplasm (Minns et al., 2018). Phosphorylation of initiation factors such as eIF2-α, eIF4E, and Poly-Adenylate Binding Protein is an important mechanism of regulating mRNA translation (Hay and Sonenberg, 2004). For instance, the phosphorylation of poly A-binding proteins (PABP) improves its interaction with eIF4G and enhances the rate of mRNA translation (Le et al., 2000).
PABP is a post-transcriptional RNA binding protein which binds to the adenine-rich sequences in mRNA and acts as a scaffold for protein-protein interactions (Mangus et al., 2003). In model eukaryotes, translationally repressed proteins interact with and are influenced by PABPs. In the nucleus, PABP play a role in polyadenylation; it determines the length of the poly(A) and may be involved in mRNA export (Minns et al., 2018). While in the cytoplasm, they participate in the regulation of translation initiation and either protect mRNAs from decay through binding to their poly(A) tails or stimulate this decay by promoting mRNA interactions with deadenylase complex proteins (Eliseeva et al., 2013). Non-nuclear PABP plays a surprise role outside the parasite's cell. For example, when the parasite develops into a sporozoite in the mosquito, PABP accumulates at the surface of the sporozoite and is shed when the parasite moves (Minns et al., 2018). The accumulation of this protein on the surface of the sporozoite may be involved in the binding of exogenous RNAs that accumulate as stress granules or help the parasite interact with its environment (host or vector) (Mair, 2013; Minns et al., 2018). PABP also plays a role in the interaction of poly(A) tail with translation initiation complex, and subsequent binding of eukaryotic initiation factor 4E (eIF4E) and PABP to eIF4G, which is pertinent for efficient translation (De Gregorio et al., 1999; Mangus et al., 2003; Preiss and Hentze, 2003). Using bioinformatics tools, P. falciparum PABP has been characterized and shown to have the characteristics of a cytosolic PABP with additional sequence inserted between RNA-recognition motif (RRM) III and IV (Tuteja, 2009; Tuteja and Pradhan, 2009).
It is important to regulate the expression of PABP because of the multiple roles it plays in mRNA metabolism and stored PABP mRNA is believed to be activated by growth stimuli (Bag and Bhattacharjee, 2010). Hence, factors or agents that can interfere with the functions or expression of this RNA binding protein may disrupt the parasite life cycle and result in processes such as premature hepatic sporozoite formation. For example, studies have shown that Ik2 kinase knockout sporozoite lacks phosphorylated eIF2α (which keeps mRNAs in a translationally repressed state) and the parasites develop prematurely into the liver stage and lose its infectivity (Zhang et al., 2010). Understanding the malaria parasite’s biology and the functions of its proteins may help provide insights that would be useful in antimalarial drug discovery. In other to eliminate malaria, innovative malaria intervention strategies aimed at the transmission stages of the parasite would play a pertinent role. Though the mechanism of PABP has been reported (Bag and Bhattacharjee, 2010; Eliseeva et al., 2013), detailed computational studies to validate the chemical and structural properties, as well as compounds that may interfere with the functions of Plasmodium falciparum PABP is yet to be elucidated. In this study, we predicted the structure of P. falciparum PABP using homology modeling and also carried out active site prediction and docking simulation to provide insights on this protein as a potential antimalarial drug target.
Sequence acquisition
The amino acid sequence of PABP of P. falciparum (Accession No: ABL63812.1) was obtained from the National Center for Biotechnology Information (NCBI); a comparison of this sequence with those of RCSB-PDB (Protein Data Bank) was performed.
Physiochemical characterization
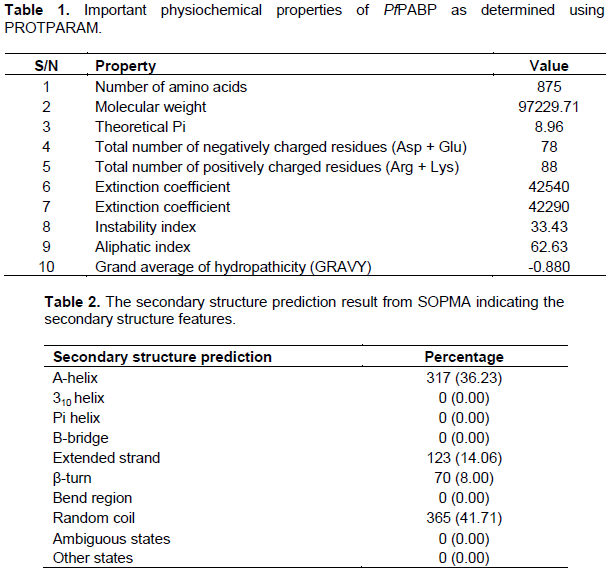
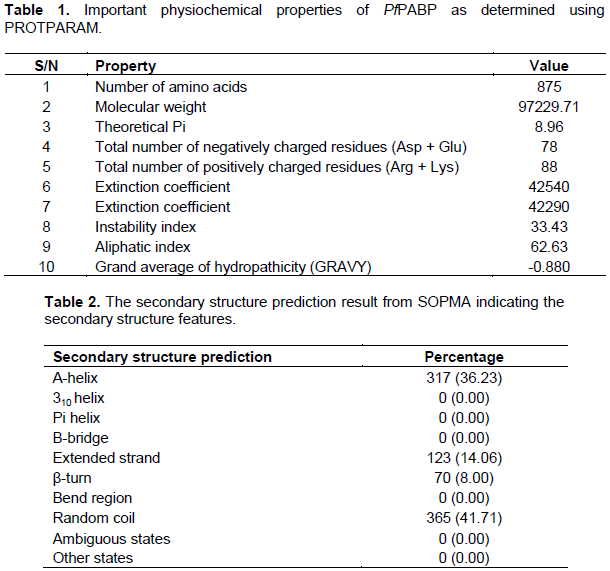
The physiochemical parameters were computed using PROTPARAM, which includes amino acid composition, molecular weight, theoretical pl, total number of negatively charged residues (Asp + Glu), total number of positively charged residues (Arg + Lys), extinction coefficient, estimated half-life, instability index and grand average of hydropathicity (GRAVY). These parameters are essential for studying protein physiochemical properties (Gasteiger et al., 2003).
Secondary structure prediction
The secondary structure of the P. falciparum PABP was predicted using the SOPMA tool (Geourjon and Deléage, 1995). This was done by making a consensus prediction from multiple sequence alignments. The positional possibility of the α-helix, β-strands, turns, and random coils of the P. falciparum PABP was assessed using default parameters with a window width of 17, number of states of 4 and similarity threshold of 8.
Network interaction
STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) was used to identify protein-protein interactions (Szklarczyk et al., 2018). STRING is a biological database and web resource for constructing different known and predicted protein interactions networks based on functional association.
Residue Interaction Network Generator (RING) was used to analyze the residue-residue interaction of Polyadenylate-Binding Protein (Piovesan et al., 2016).
Disease-causing region prediction
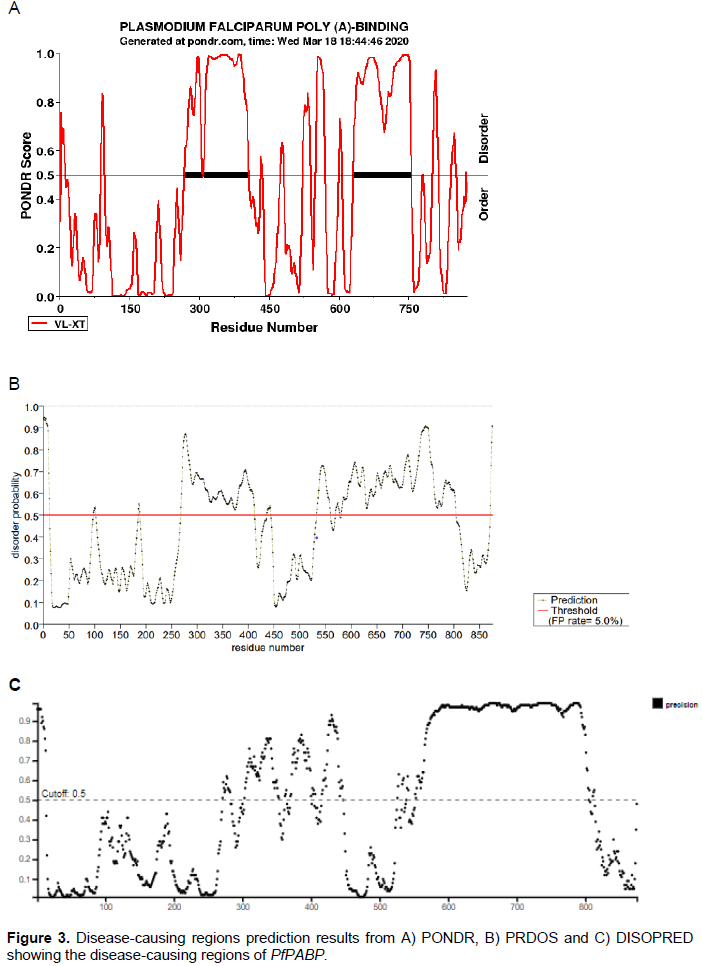
Disease-causing region prediction using PONDR, DISOPRED, and PRDOS web-based servers was analyzed to find out the regions that are intrinsically disordered in the protein. These web services look for order/globularity or disorder tendency in the query protein based on a running sum of the propensity for an amino acid to be in the ordered or disordered state by searching domain databases and known disorders in proteins.
Selection of template
Template protein is selected on sequence similarity bases. The template was searched against the PDB database using the protein-Basic Local Alignment Search Tool (BLASTp). Using the default parameters, that is, BLOSUM 62 matrix, word size 3, and an E-value threshold 10. Chain D. PABP of Saccharomyces cerevisiae (PDB ID: 6R5K_D) at a resolution of 4.8 Å sharing 51.54% identity with P. falciparum PABP was selected for structural modeling.
Structural prediction and validation
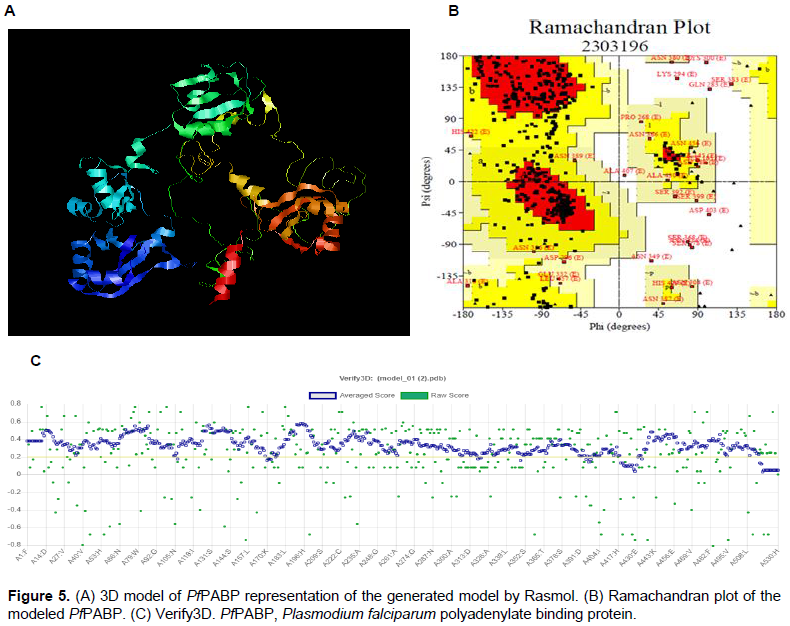
Modeling of the 3D structure of P. falciparum PABP was performed by three web-based homology modeling programs, SWISS-MODEL, PHYRE2 and, I-TASSER to compare for accuracy. The derived models were visualized using RASMOL. PROCHECK was used to check for the quality of the modeled 3D structure of P. falciparum PABP. For structure validation, the .pdb file of the modeled P. falciparum PABP was uploaded on the PDB sum web server 3.0 of European Bioinformatics Institute (EBI) to obtain both the Ramachandran plot and the Ramachandran plot statistics. While the Ramachandran plot is used in accessing the quality of a modeled protein or an experimental structure, the Ramachandran plot statistics provide information on the total number of amino acid residues found in the favorable, allowed and disallowed regions. Also, Verify3D was used to validate the structure of the modeled protein, determine how compatible a 3D structure is to its amino acids, and compare the result with that of known structures.
Binding site identification
3DLigand was employed for the identification of binding sites in the derived structure that might be responsible for interaction with eIF4G (Wass et al., 2010).
Sequence acquisition
P. falciparum PABP has been identified as an important post-transcriptional component of the organism especially in RNA metabolism and in the translational repression involved in the regulation of the parasite’s growth, development and transmission. Hence, it is an attractive target candidate for antimalarial drug discovery. The amino acid sequence of PABP of P. falciparum 3D7 with accession number ABL63812.1 was obtained from the NCBI protein database in FASTA format. The protein has a total of 875 aa with a molecular weight of 97 kD. Interpro database analysis suggests that the protein consists of RNA recognition motif domains with 16-527 aa; four Nucleotide-binding Alpha-Beta domain superfamily with 5-103, 104-177, 183-275 and 354-537 aa, Polyadenylate-binding protein/hyperplastic disc protein domain (PABP-HDP) with 799-875 aa found at the conserved C-terminal domain of the protein and functions to recruit several different translational factors to the mRNA poly (A) tail.
Physicochemical characterization
Physicochemical characterization of the protein sequence was done using the Expasy PROTPARAM tool (Gasteiger et al., 2003)to gain an insight into the PABP (Table 1). The analysis revealed an instability index of 33.43, indicating that the protein will be stable in vitro because proteins with values over 40 are considered to be unstable (Anayet et al., 2011). A low GRAVY value of -0.880 reflects the hydrated state of the protein and a high aliphatic index, calculated as the total volume occupied by the aliphatic side chains, is considered a positive physicochemical factor for increased thermostability. The high extinction coefficient also points to the stability of the protein.
Secondary structure prediction
The secondary structural features indicate whether a given amino acid lies in α-helix, β-strand, or random coil. SOPMA (self-optimized prediction method with alignment) servers was used for secondary structure prediction and the features showed domination of random coils 41.71% followed by α-helix (Hh) 36.23%, extended strand (Ee) 14.06%, and β-turn (Tt) 8.00% (Table 2). The abundance of random coils could be important in the formation of the protein’s 3D structure and also indicates a high level of stability and conservation of the protein structure (Ullah et al., 2012). Interactions between side chains within a random coil sometimes lead to the formation of hydrophobic clusters which acts as initiation or nucleation sites for protein folding (Nain et al., 2020; Smith et al., 1996). It has also been shown that random coils act as ‘connecting bridges’ for the alpha-helix and beta-strands, with the amino acid content of the random coils depending on the flanking structures (Khrustalev et al., 2013, 2014).
Network analysis
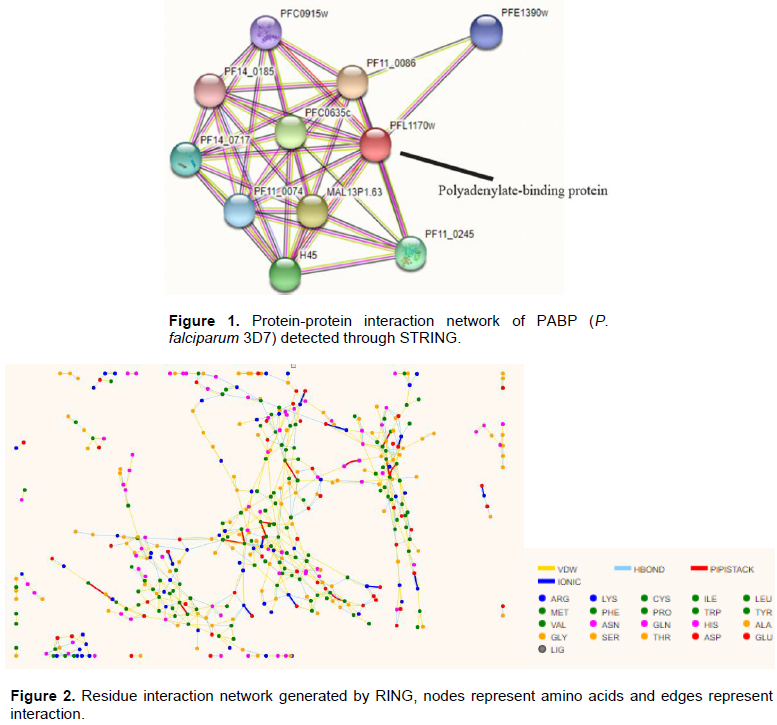
The protein-protein interaction (PPI) of PABP of P. falciparum with other proteins was determined using STRING (Szklarczyk et al., 2018). PPIs are very indicative of certain events in a cell and usually form the basis for several transcriptional regulatory networks and help elucidate signal transduction pathways in a cell (Raman, 2010). PPI of P. falciparum PABP generated through STRING is presented in Figure 1. The PPI network result shows that PfPABP interacts with other proteins in a high confidence score, among which eukaryotic initiation factors EIF4A, EIF4E, and EIF4G, members of the multi-subunit translation initiation complex EIF4F were identified to interact with P. falciparum PABP (represented as PFL1170w on the STRING result).
EIF4A (H45) is an ATP-dependent RNA helicase involved in unwinding of the inhibitory secondary structures present in the 5’-UTR of the mRNA and also aids in the binding and scanning of the ribosomes for the initiator codon due to its single-stranded form (Rogers et al., 2001). EIF4E (PFC0635c) functions in the recognition of the mRNA 5’-cap structure and also necessary for cap-dependent translation (Gingras et al., 1999). EIF4G (MAL13P1.63) is an adapter protein that is required to mediate ribosome recruitment as well as circularization of the mRNA via interaction with PABP (Tuteja, 2009).
Other proteins identified to have a network PPI interaction with P. falciparum PABP includes the ATP-dependent RNA helicases DDX6, SPB4 and DDX41 (PFC0915w, PF14_0185, and PFE1390w); the PABP-interacting protein 1 (PF11_0086) which mediates binding between PABP and EIF4A (Minns et al., 2018); the translation elongation factor subunit alpha (PF11_0245), etc.
To analyze residue interaction within the P. falciparum PABP molecule, the residue interaction network was generated by RING (Piovesan et al., 2016). This describes the protein’s 3D structure as a graph where nodes represent residues and edges represent physiochemical characterization (Anayet et al., 2011). RING uses the standard programs to create network interactions. The residue-residue interaction network of PABP indicates the probable active site of the protein (Figure 2).
Disease-causing region prediction
The results from the 3 servers showed the regions that are intrinsically disordered in the protein (Figure 3). Disordered regions of proteins are important and necessary for performing many functions such as DNA binding, binding to other proteins such as to kinases, transcription factors and translation inhibitors or mRNAs (Anayet et al., 2011; Dunker et al., 2002; He et al., 2009). These disordered regions might contain functional sites or linear motifs such as molecular recognition domains, protein folding inhibitors, flexible linkers, etc. (Dunker et al., 2001). This correlates with the results from the Interpro prediction of the various domains found in the P. falciparum PABP as well as the results from the protein-protein interaction which predicted that the P. falciparum PABP has a network of PPI with other proteins involved in binding PABP and EIF4A as well as with other proteins.
Template selection
To find a suitable template for the protein model based on sequence similarity, a BLASTp search was conducted using default parameters. From the result, Chain D PABP of Saccharomyces cerevisiae (PDB ID: 6R5K_D) at a resolution of 4.8 Å, sharing 51.54% identity with P. falciparum PABP was selected for the structural modeling.
Structure prediction and validation of modeled protein
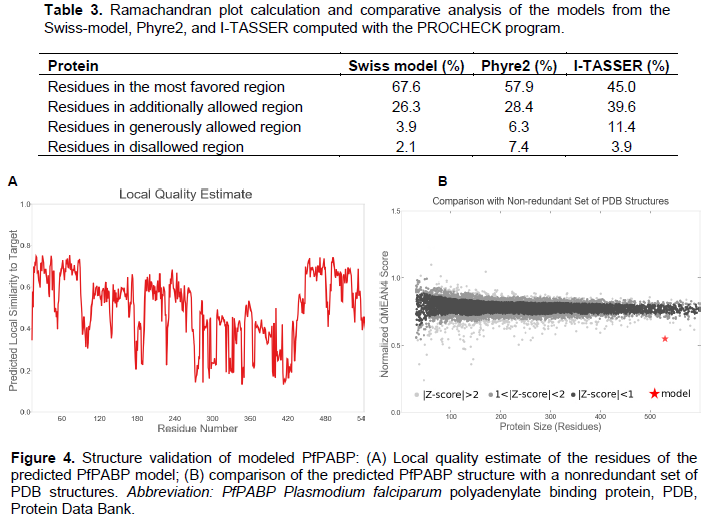
The three-dimensional structure of P. falciparum PABP performed by three homology modeling programs generated a refined 3D homology model of the protein sequence based on the given sequence alignment and the selected template (Table 3). 3D protein structures provide insight into the function of a protein since protein sequences with >20% identity may have identical structure and function. The SWISS-MODEL model web server which automatically calculates the QMEAN scoring function of the protein model, producing a z-score ranging from 0 to 1 (Arnold et al., 2006; Bordoli et al., 2009)was able to generate a better-quality 3D model with a GMQE of 0.32 and QMEAN of -6.5 (Figure 4). Ramachandran plot was done by PROCHECK to measure the accuracy of the modeled protein. About <70% of the residues were in the favored region and 2.1% of amino acids in the disallowed region (Figure 5). This also validates the modeled structure as a good quality protein model.
Binding site identification
3DLigand predicted 9 binding sites in the modeled structure. Out of which the largest site having a volume of 233 Å3 was selected as the active site. Important residues identified in the active site were ASN14, ARG 49, ASP 50, SER 51, THR 53, ARG 76, LYS 55, ARG 55, HIS 85, GLN104 and ARG167. These residues located at the RRM1 and 2 regions of the modeled PfPABP have previously been reported to interact with eIF4G in animals and yeast (Imataka et al., 1998; Kessler and Sachs, 1998).
The setback in the war against malaria is caused mainly by the increasing number of vectors resistant to insecticides, dirty environments, limited primary healthcare facilities as well as lack of affordable cheap/effective drugs and the spread of multidrug resistance Plasmodium species. These situations have strengthened the need for search of new drug targets and understanding the basic biology of the malaria parasite. Hence, our findings would help broaden understanding of P. falciparum PABP, in particular, understanding its structure, binding sites, conformational changes, protein-protein/residue interaction, diseases causing regions as well as its physicochemical properties. These data would be valuable in structure-function interventions and identifying molecular targets for designing drugs applicable to P. falciparum. The authors have identified P. falciparum PABP as a potential antimalarial drug target and they recommend in vitro and in vivo experimentation to further justify its potential.
The authors have not declared any conflict of interests.
REFERENCES
|
Anayet HM, Habibul HMM, Sultana CA, Datta A, Arif KM (2011). Molecular-docking study of malaria drug target enzyme transketolase in Plasmodium falciparum 3D7 portends the novel approach to its treatment. Source Code for Biology and Medicine 10:7.
|
|
|
|
Arnold K, Bordoli L, Kopp J, Schwede T (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics (Oxford, England) 22(2):195-201.
Crossref
|
|
|
|
|
Bag J, Bhattacharjee RB (2010). Multiple levels of post-transcriptional control of expression of the poly (A)-binding protein. RNA Biology 7(1):5-12.
Crossref
|
|
|
|
|
Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T (2009). Protein structure homology modeling using SWISS-MODEL workspace. Nature Protocols 4(1):1-13.
Crossref
|
|
|
|
|
Bunnik EM, Batugedara G, Saraf A, Prudhomme J, Florens L, Le Roch KG (2016). The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biology 17:147.
Crossref
|
|
|
|
|
Coulson RMR, Hall N, Ouzounis CA (2004). Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Research 14(8):1548-1554.
Crossref
|
|
|
|
|
Cui L, Fan Q, Li J (2002). The malaria parasite Plasmodium falciparum encodes members of the Puf RNA-binding protein family with conserved RNA binding activity. Nucleic Acids Research 30(21):4607-4617.
Crossref
|
|
|
|
|
Cui L, Lindner S, Miao J (2015). Translational regulation during stage transitions in malaria parasites. Annals of the New York Academy of Sciences 1342(1):1-9.
Crossref
|
|
|
|
|
De Gregorio E, Preiss T, Hentze MW (1999). Translation driven by an eIF4G core domain in vivo. The EMBO Journal 18(17):4865-4874.
Crossref
|
|
|
|
|
Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z (2002). Intrinsic disorder and protein fluctuations. Biochemistry 41(21):6573-6582.
Crossref
|
|
|
|
|
Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang CH, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z (2001). Intrinsically disordered protein. Journal of Molecular Graphics and Modelling 19(1):26-59.
Crossref
|
|
|
|
|
Eliseeva IA, Lyabin DN, Ovchinnikov LP (2013). Poly(A)-binding proteins: Structure, domain organization, and activity regulation. Biochemistry (Moscow) 78(13):1377-1391.
Crossref
|
|
|
|
|
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003). ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Research 31(13):3784-3788.
Crossref
|
|
|
|
|
Geourjon C, Deléage G (1995). Sopma: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11(6):681-684.
Crossref
|
|
|
|
|
Gingras AC, Raught B, Sonenberg N (1999). eIF4 Initiation Factors: Effectors of mRNA Recruitment to Ribosomes and Regulators of Translation. Annual Review of Biochemistry 68(1):913-963.
Crossref
|
|
|
|
|
Hay N, Sonenberg N (2004). Upstream and downstream of mTOR. Genes and Development 18(16):1926-1945.
Crossref
|
|
|
|
|
He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK (2009). Predicting intrinsic disorder in proteins: An overview. Cell Research 19(8):929-949.
Crossref
|
|
|
|
|
Imataka H, Gradi A, Sonenberg N (1998). A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. The EMBO Journal 17(24):7480-7489.
Crossref
|
|
|
|
|
Kessler SH, Sachs AB (1998). RNA Recognition Motif 2 of Yeast Pab1p Is Required for Its Functional Interaction with Eukaryotic Translation Initiation Factor 4G. Molecular and Cellular Biology 18(1):51-57.
Crossref
|
|
|
|
|
Khrustalev VV, Barkovsky EV, Khrustaleva TA (2014). The Influence of Flanking Secondary Structures on Amino Acid Content and Typical Lengths of 3/10 Helices. International Journal of Proteomics 2014:1-13.
Crossref
|
|
|
|
|
Khrustalev VV, Khrustaleva TA, Barkovsky EV (2013). Random coil structures in bacterial proteins. Relationships of their amino acid compositions to flanking structures and corresponding genic base compositions. Biochimie 95(9):1745-1754.
Crossref
|
|
|
|
|
Le H, Browning KS, Gallie DR (2000). The phosphorylation state of poly(A)-binding protein specifies its binding to poly(A) RNA and its interaction with eukaryotic initiation factor (eIF) 4F, eIFiso4F, and eIF4B. Journal of Biological Chemistry 275(23):17452-17462.
Crossref
|
|
|
|
|
Mair GR (2013) Translation and Its Control. In: Hommel M, Kremsner P (eds.), Encyclopedia of Malaria. Springer pp. 8757-8759.
Crossref
|
|
|
|
|
Mangus DA, Evans MC, Jacobson A (2003). Poly(A)-binding proteins: Multifunctional scaffolds for the post-transcriptional control of gene expression. In Genome Biology 4(7):223.
Crossref
|
|
|
|
|
Minns AM, Hart KJ, Subramanian S, Hafenstein S, Lindner SE (2018). Nuclear, Cytosolic, and Surface-Localized Poly(A)-Binding Proteins of Plasmodium yoelii. MSphere 3(1):1-13.
Crossref
|
|
|
|
|
Nain Z, Kumar AU, Abdulla F, Hossain N, Chandra BN, Jasin MF, Azakami H, Minnatul KM (2020). Computational prediction of active sites and ligands in different AHL quorum quenching lactonases and acylases. Journal of Biosciences 45(26):25-43.
Crossref
|
|
|
|
|
Piovesan D, Minervini G, Tosatto SCE (2016). The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Research 44:367-374.
Crossref
|
|
|
|
|
Preiss T, Hentze MW (2003). Starting the protein synthesis machine: eukaryotic translation initiation. BioEssays 25:1201-1211.
Crossref
|
|
|
|
|
Raman K (2010). Construction and analysis of protein-protein interaction networks. BioMed Central Automated Experimentation 2(1):2.
Crossref
|
|
|
|
|
Reddy BN, Shrestha S, Hart KJ, Liang X, Kemirembe K, Cui L, Lindner SE (2015). A bioinformatics survey of RNA-binding proteins in Plasmodium. BMC Genomics 16(1):890.
Crossref
|
|
|
|
|
Rogers GW, Lima WF, Merrick WC (2001). Further characterization of the helicase activity of eIF4A. Substrate specificity. Journal of Biological Chemistry 276(16):12598-12608.
Crossref
|
|
|
|
|
Smith LJ, Fiebig KM, Schwalbe H, Dobson CM (1996). The concept of a random coil : Residual structure in peptides and denatured proteins. Folding and Design 1(5):R95-R106.
Crossref
|
|
|
|
|
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Jensen LJ, Von Mering C (2018). STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 47:607-613.
Crossref
|
|
|
|
|
Tuteja R (2009). Identification and bioinformatics characterization of translation initiation complex eIF4F components and poly(A)-binding protein from Plasmodium falciparum. Communicative and Integrative Biology 2(3):245-260.
Crossref
|
|
|
|
|
Tuteja R, Pradhan A (2009). Isolation and functional characterization of eIF4F components and poly(A)-binding protein from Plasmodium falciparum. Parasitology International 58(4):481-485.
Crossref
|
|
|
|
|
Ullah M, Hira J, Ghosh T, Ishaque N, Absar N (2012). A bioinformatics approach for homology modeling and binding site identification of triosephosphate isomerase from Plasmodium falciparum 3D7. Journal of Young Pharmacists 4(4):261-266.
Crossref
|
|
|
|
|
Wass MN, Kelley LA, Sternberg MJE (2010). 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acid Research 38(2):W469-W473.
Crossref
|
|
|
|
|
World Health Organization (WHO) (2019).World malaria report 2019. Retrieved February 13, 2020. Available at:
View
|
|
|
|
|
Zhang M, Fennell C, Ranford-Cartwright L, Sakthivel R, Gueirard P, Meister S, Caspi A, Doerig C, Nussenzweig RS, Tuteja R, Sullivan WJ, Roos DS, Fontoura BMA, Ménard R, Winzeler EA, Nussenzweig V (2010). The Plasmodium eukaryotic initiation factor-2α kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. Journal of Experimental Medicine 207(7):1465-1474.
Crossref
|
|