Temperature programmed reduction (TPR) is one of the techniques for obtaining information about phases or bulk species in heterogeneous catalysts. Information from TPR analysis can give insights about phase-support interaction and extent of reduction of the phases at different temperatures. TPR technique is a common tool in the characterisation of cobalt based Fischer-Tropsch (FT) catalysts. However, interpretation of TPR profiles of γ-alumina supported cobalt FT catalysts had been characterised with different views on the nature of phases and reduction processes involved. In this report, we use reduction behaviour of unsupported Co3O4 to gain insight for more explicit analysis of TPR profiles of γ-alumina supported Co3O4 catalysts. The transition Co3O4 → CoO → Co in γ-alumina supported catalysts prepared with wet impregnation with aqueous cobalt nitrate and calcined at temperatures ≤ 350°C gave reduction peaks at 300 to 350°C. Reduction peaks at 500 to 600°C were due to Co-Al mixed oxide phases; most likely Co2AlO4 and probably routes formation of the mixed oxide were also discussed. Consideration of tendency of dissolution of γ-alumina during the impregnation of metal salt is instructive toward achieving higher reducibility of Co3O4 in the design of cobalt based Fischer-Tropsch catalysts.
Increasing energy demand coupled with the awareness of declining petroleum reserves has motivated renewed research and commercial interest in Fischer–Tropsch synthesis (FTS). Different feedstock options (natural gas, coal and biomass) are being explored to produce liquid hydrocarbons as alternative to petroleum (Kagan et al., 2008; Schulz, 1999; Dry, 2002). Commercial FTS operations are based on iron and cobalt based catalysts. H2/CO ratio of syngas from natural gas do not required water gas shift step which matches the low water-gas shift (WGS) activity of cobalt based catalysts. Moreover, Co-based catalysts are selective towards paraffins and have higher activity for hydrocarbon formation than Fe-based catalysts; hence, cobalt catalysts are preferred for gas-to-liquid (GTL) projects (Perego, 2007; Schulz, 1999; Schulz, 2003).
Active sites in cobalt based catalysts are cobalt metal nanoparticles and several studies have shown positive correlations between cobalt dispersion and FTS activity and hydrocarbon selectivity (Iglesia, 1997; Girardon et al., 2007). Thus, maximizing cobalt dispersion on the support is a key objective in the design of Co-based catalysts (Khodakov, 2009). Generally, preparations of cobalt catalysts involve impregnation of high surface area support material with cobalt salt solution, followed by drying and calcination. During calcination, the impregnated salt is transformed into oxide phase(s). The active phase for the FT synthesis are generated in situ by reduction under hydrogen stream prior to passage of syngas feed (Zhang et al., 2002; Sirijaruphan et al., 2003).
Gamma (γ) alumina is often a prefer support for cobalt catalysts because of its ability to stabilize small size clusters of cobalt particles and high resistance to attrition especially in the continuously stirred tank reactor or slurry bubble column reactor. However, several reports have advanced that after activation, considerably large fraction of the impregnated cobalt precursor on γ-alumina are catalytically inactive for the hydrocarbon synthesis (Chu et al., 2007; Sirijaruphan et al., 2003). An important of objective in the design of cobalt-based Fischer-Tropsch synthesis is to maximise the proportion of impregnated cobalt precursor on γ-alumina that become reduced to the metallic phase. Temperature programmed reduction (TPR) technique is useful barometric tool for studying the reduction behaviour of catalysts. However, there are varied interpretations of H-TPR profiles of γ-alumina supported FTS cobalt catalysts. These interpretations was categorised and summarised by Jacobs et al., (2007) (Figure 1). Case 1 scenario is the most popular, with the view that the predominant phase of the cobalt is as Co3O4 and it undergo reduction to metallic cobalt in a two stage process: Co3O4 → CoO → Co, at temperatures ranges 200 to 400°C and 400 to 800°C, respectively. Case 2 and Case 3 scenarios recognised Co3O4 phase with a one-stage reduction to Co, at the temperature ~ 300 to 350°C. The advocates of the Case 2 and Case 3 viewpoints believed that γ-alumina supported FTS cobalt catalysts also contain CoO or xCoO-yAl2O3 cobalt phases. It is argued that reduction of these phases account for H-TPR peaks in the temperature range of 400 to 800°C. The three proposals recognised the possibility of cobalt-aluminate phase which is reduced above 800°C.
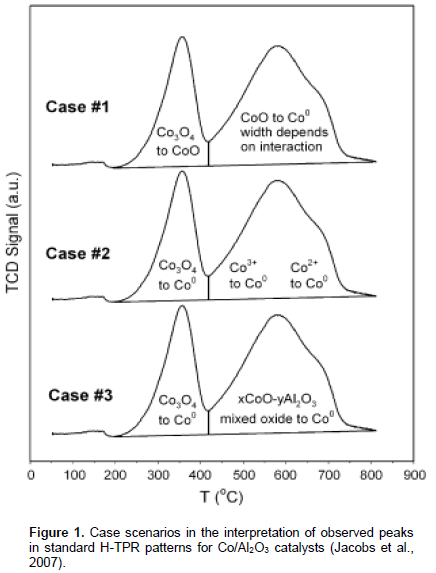
While there is yet a consensus on reduction behaviour of alumina supported cobalt catalyst, the aforementioned highlighted perspectives in the interpretations of H-TPR profiles have influenced approaches to design of alumina supported cobalt based FT catalysts. This study is motivated by the need for correct interpretations of H-TPR profiles alumina supported cobalt catalysts that are intended for Fischer-Tropsch application. Hence, we present results of H-TPR studies of unsupported cobalt oxide as reference to gain insight about reduction behaviour of γ-alumina cobalt catalysts. X-ray diffraction (XRD) was also used to identify the Co-oxide phases and determine their crystallite sizes. Reducibility of the supported catalysts was determined by O2-titration. Estimates of percentage reduction of the catalysts were also made from the TPR profiles. Result of this study does not agree with two stage process (Case 1; Co3O4 → CoO → Co) for conversion of cobalt oxide to metallic cobalt. It shows that design of alumina supported cobalt catalysts should be approached with the view of cobalt oxide reduction following the Case 3 scenario.
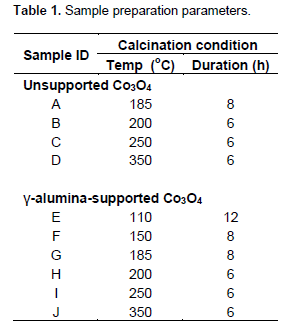
Catalyst preparation
Unsupported Co3O4 samples were prepared by calcining cobalt nitrate hexahydrate [Co(NO3)2.6H2O, Merck, India] at 185, 200, 250 and 350°C for 8, 6, 6 and 6 h, respectively. Gravimetric measurements of their thermal decomposition in static air were carried out by weighing 5 g of the salt into pre-weighed clean alumina crucibles. After calcination, the crucible and the contents were cooled in a desiccator, re-weighed and the mass loss recorded. Single lot of 60 g of supported cobalt catalyst is prepared by wet impregnation method with cobalt loading (20 mol %) on γ-alumina (Sasol GmbH, Germany, Extrudates, 1.6/200, Lot: E317, Spec.: 665100, bulk density 0.73 g/cm3; BET Surface Area 181 m2g-1; Pore Volume: 0.49 cm3g-1). The sample was dried for 12 h at 110°C and divided into six equal portions. Details of calcination of the catalysts are presented in Table 1.
X-ray diffraction
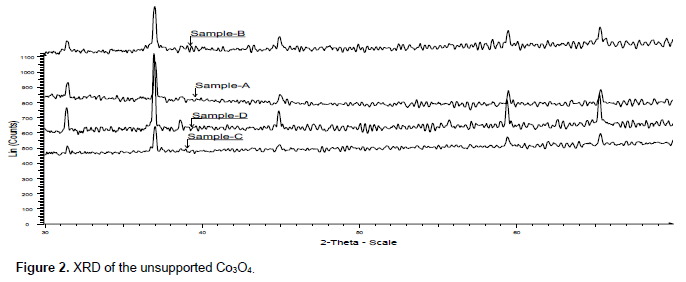
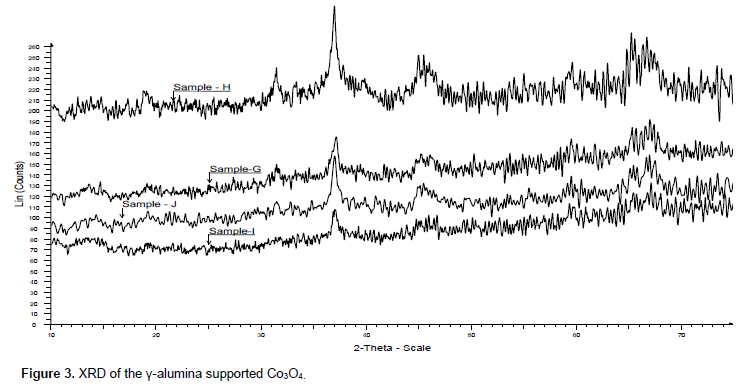
XRD patterns were recorded at room temperature on a D8 ADVANCE (BRUKER AXS, Germany) diffractometer using CuKα radiation with parallel beam (Gobel Mirror). The catalysts were ground to fine powder prior to measurement. The scans were recorded in the 2θ range between 10 and 75° using step size of 0.02° and scan speed of 2 s/step. Peaks were identified by search match technique using DIFFRACplus software (BRUKER AXS, Germany) with reference to the JCPDS database. The software TOPAS 3.0 from Bruker AXS (2005) was used for refinement of Co3O4 diffraction peak (311) located at 2θ = 36.9° to determine the average crystallite size.
Temperature programmed reduction (TPR)
TPR profiles of the samples were recorded with ChemiSorb 2720 (M/s Micrometrics, USA) equipped with a TCD detector. The TPR profiles were obtained by reducing the catalyst samples by a gas mixture of 10% H2 in Ar with a flow rate of 20 ml/min while the temperature was increased from ambient to 800°C at a rate of 10°C/min.
Gravimetric measurements of thermal decomposition
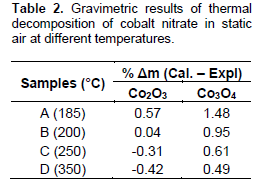
It has been reported that thermal decomposition of cobalt nitrate to cobalt oxides (Co2O3 and Co3O4) in an inert atmosphere starts at 185°C (Ehrhardt et al., 2005). This informed the choice of lowest calcination temperature for unsupported cobalt catalysts for this study. Result of the gravimetric measurements of thermal decomposition of cobalt nitrate in static air is presented in Table 2. Due to possibility presence of residual nitrate due to incomplete decomposition of the nitrate salt we anticipated that the calculated value will be higher than the experimental mass loss. Table 2 showed difference between percentage calculated and experimental mass loss of decomposition of Co(NO3)2.6H2O decreases with increasing calcination temperature.
Crystallite size
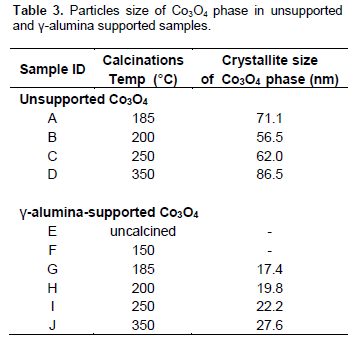
XRD of unsupported and γ-alumina supported catalysts also depict presence of Co3O4 phase (Figures 2 and 3). This corroborates the inference from the gravimetric analysis. Crystallite size of the Co3O4 of the catalysts calcined at different temperature is presented in Table 3. In both unsupported and γ-alumina supported catalysts crystallites size of Co3O4 increases with increase in calcination temperature except the unsupported sample calcined at 185°C which is an outlier. Reason for this outlier situation is not clear at the moment. At the same temperature Co3O4 crystallite sizes are smaller in the supported than in unsupported catalysts. This is due to stabilization of Co3O4 nanoparticles by the support and in each case the increasing crystallite size with increasing calcination temperature can be attributed to sintering which is promoted at higher temperatures.
H-TPR analysis
Unsupported Co3O4 samples
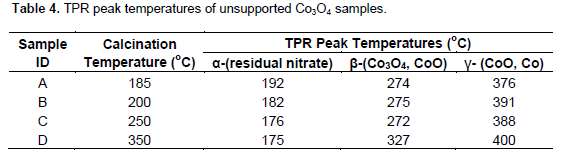
Figure 4 shows the H-TPR profiles of the unsupported Co3O4 catalysts. The temperature ranges of the peaks in the profiles are > 200; 270 to 280; 319 to 326 and 375 to 400°C. The peaks are assigned to three transitions presented in Table 4. The first hydrogen consumption temperature range is attributed to reduction of residual nitrate. The second and the third ranges assigned to Co3O4 → CoO; while the fourth is ascribed to CoO → Co. The profiles display similar trends with those reported by Chen et al., (2003), for CoOx (1.00 < x < 1.33) nanoparticles where it was shown that the value of ‘x’ influenced pattern of H-TPR profile of CoOx; x = 1, a single peak was obtained. For x > 1, a shoulder peak developed in the profile of CoOx samples and was attributed to two stage reduction of the oxides vis - CoOx → CoO → Co. Increase in the area of the shoulder peak is proportional with the value of ‘x’. They report that the dominant reduction peak temperatures decreased as the values of ‘x’ increases. The assigned transitions also agree with the report of Yuvaraj et al., (2003). These authors investigated direct reduction of transition metal nitrates with hydrogen. They advanced that reduction of transition metal nitrates is activated by their decomposition and nitrate salts decompose at temperature range between 177 to 270°C. In particular, the temperature ranges 242 ± 30° and 242 ± 20°C was reported for decomposition and reduction of cobalt nitrate, respectively.
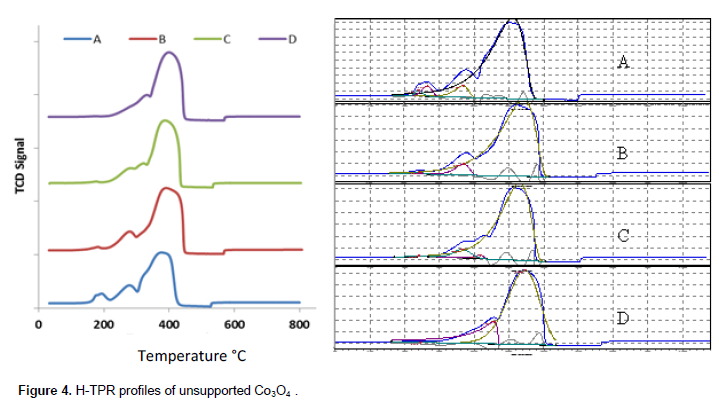
The peak temperatures assigned to reduction of residual nitrate decreases with increase in calcination temperature and are similar for catalysts C and D. Peak temperatures attributed to CoO → Co transition increases with the increase of calcination temperature and catalyst B and C also show similar temperature for CoO→ Co reduction. The trend of CoO → Co reduction peak temperature agrees with the report of Tang et al., (2008) in which it was shown that temperature of reduction of CoOx species increases with the increase of calcination temperature. Potoczna-Petru and KÈ©piÅ„ski (2001) also demonstrated that initiation temperature of reduction of Co3O4 increases with increase in calcination temperature. With the help selected area electron diffraction (SAED) and high resolution transmission electron microscopy (HRTEM) analyses the authors showed that crystallite size and morphology have strong influence on extent of reduction of Co3O4. They also demonstrated that the reduction of Co3O4 occurs via preferential epitaxial growth of CoO and Co phases on Co3O4 in course of the reduction process. Thus, reduction temperatures presented in Table 4 followed the same trend with similar studies in the literature.
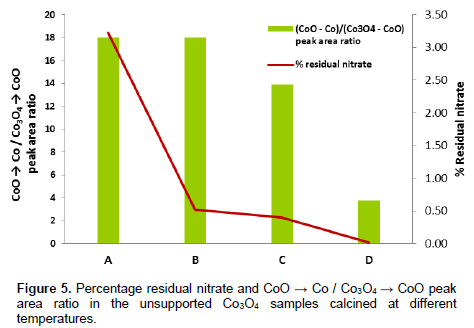
Results in Table 4 indicated that temperature gap between peaks assigned to Co3O4 → CoO and CoO → Co reduction stages narrows with increasing temperature. Deconvolution and analysis of the peak areas in the H-TPR profiles of unsupported Co3O4 samples show that the percentage residual nitrate decreases with increasing calcination temperature. The areas under the peaks assigned to residual nitrate follow similar trend with the result of the gravimetric analyses (Figure 5). The ratios of the peak area assigned to Co3O4 → CoO and CoO → Co, presented in Figure 5, suggest that the ratio varies with calcination temperature. Although the raw profile showed that the shoulders to the main peak (assigned to Co3O4 → CoO reduction stage) are more visible in samples calcinated at lower temperatures, the deconvoluted chart indicated that this (Co3O4 → CoO) reduction stage is a less distinct stage samples but its signature become more noticeable in the sample calcinated at higher temperatures. Thus, we inferred from the H-TPR profiles that reduction of Co3O4 appear to be a two stage reaction, Co3O4 → CoO→ Co, with process reduction narrow temperature gap (73 to 126°C). This conclusion corroborates the report of Chen et al., (2003) and Yuvaraj et al., (2003). It also agrees with the report of Wang et al. (2004). They also advanced that reduction of Co3O4 to metallic cobalt occurs in two stages at 160 to 230°C (Co3O4 → CoO) and 230 to 380°C (CoO → Co). Using in situ STG-TPR, these authors showed that reduction of Co3O4 and CoO is accompanied by change of shape and crystal structure: Co3O4 (hollow spheroidal shape) → face centered cubic (CoO). They added that CoO → Co transition is also associated with changes in the microstructure of the cobalt phases.
Supported Co3O4 samples
We attempted interpretation of the reduction pattern of γ-alumina supported cobalt catalysts with the backgrounds from the TPR profiles of unsupported Co3O4. Figure 6 shows the TPR profiles of the alumina supported Co3O4 samples. The TPR profiles can be deconvoluted into three peaks as presented in Table 5. The first peak (α – peak) is assigned to nitrate reduction. Compared to the unsupported catalysts, these peaks appeared at higher temperatures, which increased with increasing temperature of calcination of the supported catalysts. This may be due to interaction with the surface of the alumina support. Highest peak temperature in the unsupported Co3O4 is 400°C, and invoking crystallites effect of highest lower peak temperature of Co3O4 phase in the supported catalyst is expected to be below 400°C. However, the profiles of the supported Co3O4 contain peaks at temperature above 400°C. Moreover, the temperature gaps between the two higher temperature peaks in supported Co3O4 are much wider (191 to 436°C) than in the unsupported samples. Based on these observations we posit that the transitions in the third or γ-peaks of the unsupported and γ-alumina supported cobalt catalysts are different.
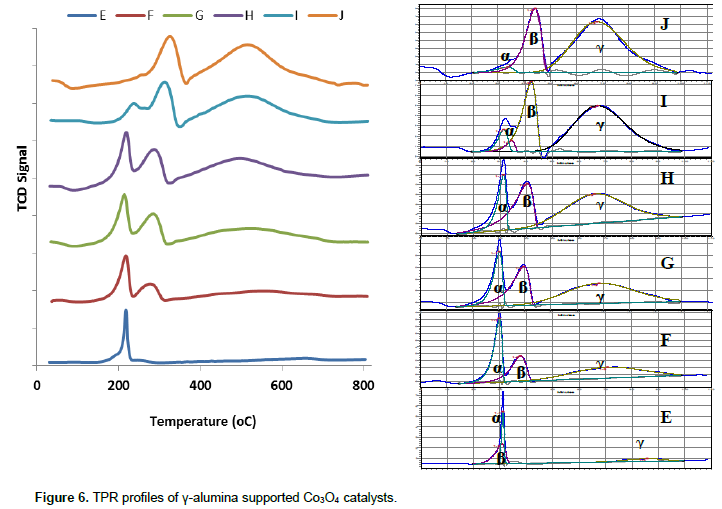
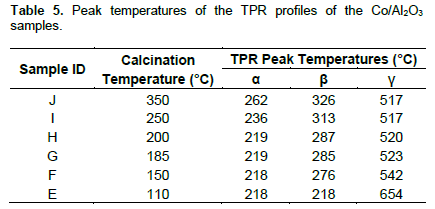
Except sample E, the second peaks in the profiles of γ-alumina supported are assigned to Co3O4 → Co transition. These peaks are at lower temperatures compared to similar transition in the unsupported counterparts. The lowered Co3O4 → Co transition temperature here is attributable to crystallites size effect in agreement with related literature reports (Potoczna-Petru and KÈ©piÅ„ski, 2001; Wang et al., 2004). The prominent peak in the uncalcined catalyst (E) consists of overlap of two peaks: nitrate and Co3O4 → Co transition peaks. The third peaks (γ – peaks) which range from 517°C (for calcined catalyst) to 654°C (for uncalcined catalyst) are designated to the reduction of cobalt-aluminium mixed oxide. The following trends are observed from the profiles of the supported catalysts: (i) temperature of nitrate (α – peak) increases but peak area decreases with increasing calcination temperature; (ii) temperature and peak area of Co3O4 → Co (β – peaks) transitions increasing with increasing calcination temperature; (iii) increasing visibility of the third (γ-peaks) with increasing calcination temperature. These third peaks are absent in the unsupported catalyst.
Dispersion and reducibility of supported cobalt catalysts is usually estimated using H2 chemisorption and O2 titration technique. Depending on cobalt loading, the reducibility values for unpromoted alumina based catalysts commonly vary between 10 to 45% (Jacobs et al., 2002; Borg et al., 2007). The temperature, 350°C, is widely adopted as the standard reduction temperature of supported cobalt catalysts for these analyses. This adopted temperature presupposes formation of metallic cobalt phase should take place at ≤ 350°C. It corroborates with the reduction temperatures obtained for unsupported Co3O4, and in effect ruled out Case# 1 scenario (Figure 1).
Results of reducibility of the supported Co3O4 catalysts are shown in Figure 7. Reducibility values obtained from H-TPR agrees well with values obtained from O2-titration method. It is observed that the reducibility determined from both the techniques has little variation of values with reference to the increase of calcination temperature. Since residual cobalt nitrate is readily transformed to cobalt during reduction in hydrogen at 350°C, the small difference may be attributed to possibility of metallic cobalt contribution from residual nitrate. The results indicated that calcination temperature has little or no influence on the reducibility of the Co/Al2O3 catalysts; crystallite size of Co3O4 phase and by extension dispersion of metallic cobalt nanoparticles in reduced catalysts decreases with increasing calcination temperature.
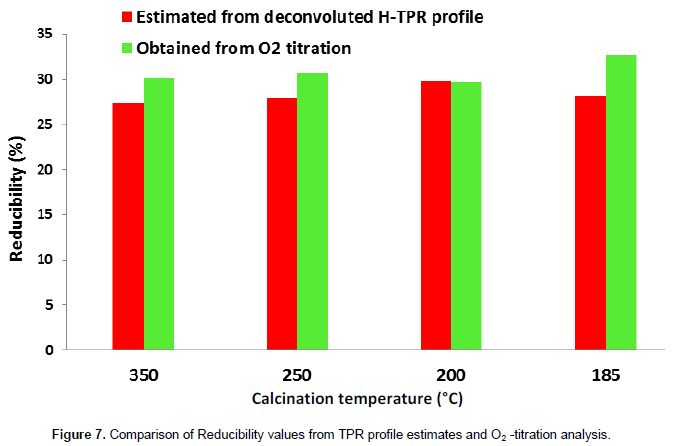
Figure 8 shows the summary of deconvolution analysis of the profiles in Figure 6. The area of the α-peaks decreases expectedly with increasing calcination temperature. It was expected that the areas of α peaks will decreased progressively from catalyst E to J. But F (calcined at 150°C) showed higher percentage hydrogen consumption than catalyst E (calcined at 110°C). According to TGA profiles of cobalt nitrate by Ehrhardt et al., (2005), calcination temperatures of the two catalysts (E and F) are below decomposition of cobalt nitrate, but may contain different amount of hydrated water molecules. The hydrated water molecules contribute to mass of the catalyst and it is expected to be higher in catalyst E than in catalyst F. Thus catalyst F will contain higher amount of cobalt nitrate per unit mass than catalyst E. This may account for the observed higher percentage hydrogen consumption production of nitrate peak of catalyst F compared to catalyst E.
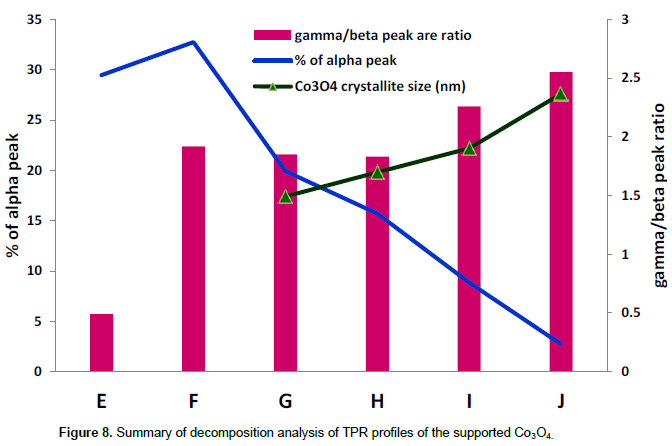
The areas of the β- and γ-peaks increase with increase in calcination temperature. The shape of the Co3O4 reduction peak is similar for both the supported and unsupported catalysts, but the signature of two stage reduction of Co3O4 particle to Co is not distinguished in the TPR profile of the supported catalysts. Due to smaller crystallite size of Co3O4 phase in the supported compared to unsupported catalysts, the two stage reduction process, Co3O4 → CoO→ Co, are combined in the β peaks. It is worth noting that there is a compromise between nitrate removal and Co3O4 crystallite in the supported catalysts. Temperature 250°C appears to be a reasonable compromise between the two ends. The same has also been suggested in the literature as optimum calcination temperature towards achieving higher cobalt dispersion on γ-Al2O3 (Borg et al., 2007; Belambe et al., 1997).
Cobalt phase in catalyst E is essentially in the nitrate form. As reported by Yuvaraj et al. (2003), concurrent decomposition and reduction of cobalt nitrate is expected at 242 ± 30°C, thus H-TPR profile of catalyst E ought not to show any peak at temperature above 350°C. However, the profiles of catalyst E showed unexpected hydrogen consumption over a broad temperature range with peak temperature at 650°C. This suggest that prior to calcination the impregnated cobalt nitrate had interacted with γ-alumina supported to form cobalt phases that are not reducible at typical reduction temperature of Co3O4 phase. While the scope of the present does not cover identification of this cobalt phase, it is important to draw attention to the point that contrary to the widely held view that γ-alumina is inert in aqueous environment, it has been shown that γ-alumina is active in aqueous media.
Carrier et al., (2007) demonstrated that hydroxide polymorphs of aluminium are thermodynamically more stable phases than γ-alumina in aqueous media. They also showed that γ-alumina transformed to hydroxide polymorphs in aqueous environment. The transformation takes place via surface hydration and dissolution. The dissolution can produce autonomous aluminium hydroxide phase from a supersaturated solution of dissolved aluminium ions. The dissolution process can also be aided by presence of H+, OH-or by metal ions (for example, Ni2+, Co2+) (Trueba and Trasatti, 2005). Thus, in effect γ-alumina in contact with aqueous metal ions solution during catalyst preparation via impregnation method has two active phases – surface hydroxide and dissolved Al3+. The surface O-Hs accounts stabilize nano clusters or particles, leading to the widely recognised high dispersion of impregnated metal of metal oxide in γ-alumina support. The second implication of alteration of γ-alumina in aqueous media to catalyst preparation is that, during evaporation stage of impregnation process it leads to supersaturation of dissolved Al3+ ions which precipitate as independent hydroxide phase or co-precipitate with metal ions being impregnated on the γ-alumina. Co-precipitation of Al3+ ions with divalent metal ions like Co2+ can form hydrotalcite-like mixed oxide phase (Ay et al., 2009).
Formation of hydrotalcites-like structures in co-precipitated of Co2+ and Al3+ ions had been reported by Khassin et al., (2001). They demonstrated that during calcination the hydrotalcites-like structures are transformed into aluminates through inverted spinel-like structure. But at moderate temperatures the hydrotalcites-like structures transformed into Co oxide phase on a highly defective inverted spinel-like structure, in which Co2+ enter the support structure and occupy both tetrahedral and octahedral positions. The authors explained that octahedron coordinated Co species are reduced at 580 to 620°C, reduction at 470 to 480°C can also yield Co° supported on inverted spinel-like structure, which contains Co2+ in the octahedral coordination. Reduction at 600°C transforms the support to ‘ideal’ spinel, which contains no octahedron coordinated Co2+. According to Pe et al., (2001), these cobalt phases may yield metallic when reduced in hydrogen at ≥ 480°C.
Thus, we suggest that the cobalt phase responsible for the γ-peaks of catalyst F-J are attributed to hydrotalcite-like structures, dehydration and dehydroxydecarbonation of these structures appear to at 150 to 200 and 250 to 300°C in air, respectively leading to inverted spinel-like structures (Co2AlO4 or CoAl2O4). In line with the discussion on alteration of γ-alumina in aqueous media, the peak at 650°C may be assigned to surface Co–Al mixed oxide phase which is formed after impregnation as amorphous phase and crystallises in situ during the course of the TPR analysis. The crystallisation of the Co–Al mixed oxide phase on calcination can also be inferred by the observed progressive sharpening of γ-peaks with increasing calcination temperature. This inference agrees with similar conclusion by Ji et al., (2000) who proposed that Co3O4 phase on Co/Al2O3 prepared by impregnation method is interfaced with other cobalt surface phase. We proposed that the cobalt phase in the third peak (γ-peak) of the calcined alumina supported catalysts is likely to be Co2AlO4 since the peak temperature is ~ 520°C which near to 480°C. But this Co2AlO4 phase could not be identified by XRD analysis due to very similar lattice parameters of the phases namely Co3O4, Co2AlO4 and CoAl2O4 (Walsh et al., 2007).
H-TPR profiles of unsupported cobalt catalysts calcined at different temperatures are used as reference for TPR profiles of the supported catalysts. The amount of residual nitrate decreases with increasing calcination temperature. Reduction Co3O4 to Co occurs in two stages: Co3O4 → CoO → Co at temperature ranges of 200 to 400°C and 220 to 330°C for unsupported and supported catalysts, respectively. The two reduction steps take place within same temperature region with single peak maximum in the supported catalysts. Calcination temperature does not have significant effect on reducibility of alumina supported Co3O4, but 250°C is considered a reasonable compromise between achieving high nitrate removal and Co3O4 dispersion. The result of this study is not consistent with the interpretation of TPR profile of phase Co3O4 on alumina support in terms of a two stage reduction process with peak temperatures at 200 to 400°C and 400 to 800°C, respectively. We suggest that consideration of the tendency of dissolution of alumina during the impregnation of metal salt will be beneficial toward achieving higher reducibility of Co3O4 in the design of cobalt based Fischer-Tropsch catalysts.
The authors have not declared any conflict of interest.
James is grateful to TWAS (Triestle, Italy) and CSIR (India) for postgraduate fellowship.