ABSTRACT
The polymerase chain reaction (PCR) is a relatively simple technique that amplifies a DNA template to produce specific DNA fragments in vitro. Basic PCR is commonplace in many molecular biology labs where it is used to amplify DNA fragments and detect DNA or RNA sequences within a cell or environment. The method is rapid, cost efficient, and when combined with suitable internal controls can be applied to the detection and quantification of specific soil organisms or pathogens on a large-scale basis. In the present study, we have adapted this approach to soil samples, providing for a simple extraction protocol which can be used directly with PCR amplification without additional DNA purification
Key words: DNA extraction, polymerase chain reaction (PCR), soil organisms.
Polymerase chain reaction (PCR) is a revolutionary method developed by Kary Mullis in the 1980s. PCR is based on using theability of DNA polymerase to synthesize new strand of DNA complementary to the offered template strand. Polymerase chain reaction (PCR) has rapidly become one of the most widely used techniques in molecular biology and for good reason: it is a rapid, inexpensive and simple means of producing relatively large numbers of copies of DNA molecules from minute quantities of source DNA material--even when the source DNA is of relatively poor quality.
The PCR is a scientific technique in molecular biology to amplify a single or a few copies of a piece of DNA across several orders of magnitude, generating thousands to millions of copies of a particular DNA sequence. PCR is used to amplify a specific region of a DNA strand (the DNA target). Most PCR methods typically amplify DNA fragments of up to ~10 kilo base pairs (kb), although some techniques allow for amplification of fragments up to 40 kb in size (Cheng et al., 1994). PCR is being applied more often to the assay of microorganisms in the environment, including soils (for a review, Steffan et al., 1991). The simplicity of this technology, together with its potential to detect small numbers of target organisms without a need for the culturing of cells, easily makes it an important method for monitoring pathogens and indicator bacteria. Despite this potential, technical limitations have continued to limit the large-scale use of PCR with soil samples primarily because extraction techniques have been laborintensive and often unreliable. While debate regarding the potential for genetic exchange in soils has continued for more than two decades and genetically engineered organisms are being released ever more frequently, the quantification of genetic transfer and our knowledge of the fate of genetic materials in soils remain surprisingly limited. Such questions underline the need to develop more-effective large-scale methods which can be efficiently applied when many samples must be evaluated. Subsequently modified the procedure by using polyvinylpyrrolidone to remove soil organic matter from the cell preparations and repetitive cesium chloride density gradient centrifugation to purify the DNA. While these approaches have been effective, they remain very labor-intensive. Methods also have been developed specifically for use with PCR amplification. For example, Pillai et al. (1991), developed a method to separate bacterial cells by modified sucrose gradient centrifugation, but again the approach is too labor-intensive for wide-scale application. The direct extraction of DNA from soil organisms without prior purification or culturing clearly would provide an attractive alternative.
Organisms and plasmids
Three types of DNA were used as targets for PCR amplification: purified Verticillium dahliae genomic DNA, an internal control template cloned in pTZ19R (Hu et al., 1993), and purified V. dahliae microsclerotia, kindly provided by G. Lazarovits. For genomic DNA, mycelia were grown without light in Czapek’s broth Tuite (1969), at 228°C with shaking, and the DNA was extracted by the hexadecyl trimethylammonium bromide (CTAB) method of Rogers and Bendich (1985), as previously described by Hu et al. (1993). The plasmid control template DNA was prepared as described by Holmes and Quigley (1981). Both types of DNA were purified further by CsCl density gradient centrifugation (Radloff et al., 1967), and the amount of DNA was determined at A260 with the assumption that 1 unit of double-stranded DNA at A260 is equivalent to 50 mg/ml.
Extraction of DNA from soil
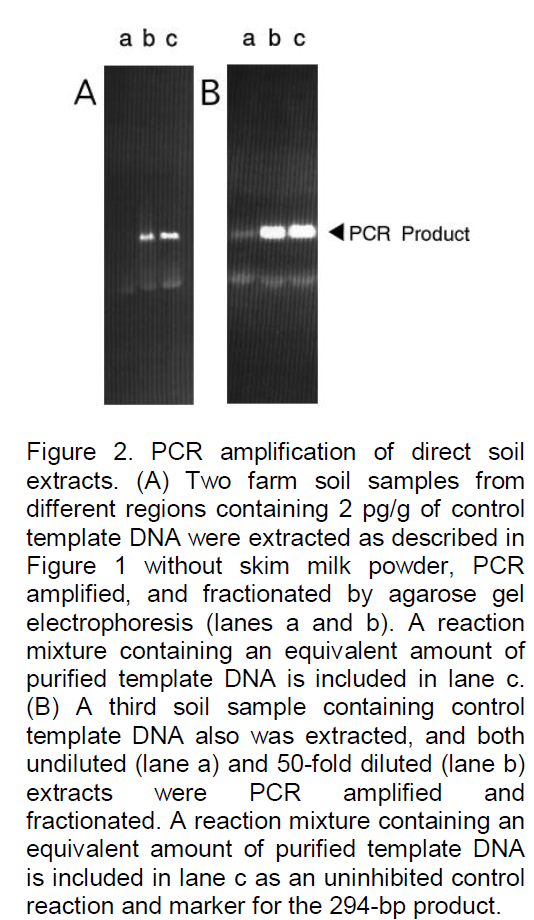
The optimized protocol developed in this study was based on previously described direct extraction methods for plant tissues containing Verticillium pathogens (Nazar et al., 1991). As indicated in Figure 1, in the basic procedure, 0.25 g of soil sample is ground with liquid nitrogen by using a mortar and pestle for about 5 min or until a fine powder remains. The powdered soil is suspended in 0.5 ml of skim milk powder solution (0.1 g of milk powder in 25 ml of H2O) by vigorous vortexing; for quantitative assays, internal control template DNA (usually 500 pg) is also added at this time. The soil and debris are removed by centrifugation at 48°C (12,000 3 g, 10 min), and the supernatant is mixed with 2 ml of sodium dodecyl sulfate (SDS) extraction buffer (0.3% SDS in 0.14 M NaCl, 50 mM sodium acetate [pH 5.1]) by vortexing. An equal volume of water-saturated phenol solution (Steele et al., 1965), is added; the phases are mixed by intermittent vortexing for 2 min at room temperature and then separated by centrifugation (12,000 3 g, 10 min).
PCR amplification of soil DNA extracts
Five microliters of DNA extract was assayed; usually, the extract was first diluted 50-fold to reduce or avoid inhibiting substances. PCR amplification normally was conducted by using 50 ml. The PCR reaction mixture containing PCR buffer (normally, 50 mM KCl, 1.5 mM MgCl2, 10 mM Tris-HCl [pH 9.0], 0.1% Triton X-100), 0.1 mg of bovine serum albumin (BSA) per ml, 0.2 mM each deoxyribonucleotide triphosphate, 12.5 pmol of each V. dahliae specific oligonucleotide primer (Nazar et al., 1991), 2 U of Taq DNA polymerase (Promega Corp., Madison, Wis.), and the DNA extract. The primers were synthesized by using a Cyclone Plus automated oligonucleotide synthesizer (Milligen/Biosearch, Milford, Mass). The amplification was performed in a programmable block (Pharmacia Biotech, Uppsala, Sweden) by using 30 reaction cycles, each consisting of a 1-min denaturation step at 948°C, a 1 min annealing step at 608°C, and a 2 min elongation step at 728°C. For nested PCR amplifications, the first amplification was carried out with a second set of oligonucleotide primers (CTCATAACCC TTTG TGAACC and CCGAGGTCAACCGTTG CCG), with target sites external to the standardized V. dahliae-specific primers which are used in the second amplification phase.
The products of PCR amplification were analyzed after fractionation by agarose gel electrophoresis. Usually, 5 ml of the PCR reaction mixture was mixed with 2 ml of loading dye (5% SDS, 25% glycerol, 0.025% bromophenol blue), heated to 658°C for 1 to 3 min, and loaded on a 2% horizontal slab gel (McDonnell et al., 1977). When the dye marker was approaching the bottom of the gel slab, the gel was stained for 40 min with ethidium bromide (0.5 mg/ml), rinsed with water (Sharpe et al., 1973), and visualized with a UV transilluminator (300 nm). For quantitative measurements, a charge-coupled device camera imaging system and Molecular Analyst/PC software (Bio-Rad Laboratories, Hercules, Calif.) were used to capture the image and to calculate the band intensities.
In this study, three potential problems were considered in the application of PCR amplification to soil samples: DNA losses due to degradation and adsorption as well as reaction inhibiting contaminants. In parallel, an attempt was made to maximize the simplicity of any extraction procedure.
Because cell or DNA purification steps are especially labor-intensive, direct extraction after cell grinding in liquid nitrogen was adapted as a minimal method for cell disruption and DNA extraction. Previous experience had indicated that grinding in liquid nitrogen was entirely sufficient to disrupt both plant and fungal tissues (Nazar et al., 1991). In the present study, the soil actually provided additional abrasion in the cell disruption process and the use of liquid nitrogen allowed cell disruption under temperature conditions which minimized nucleic acid degradation.
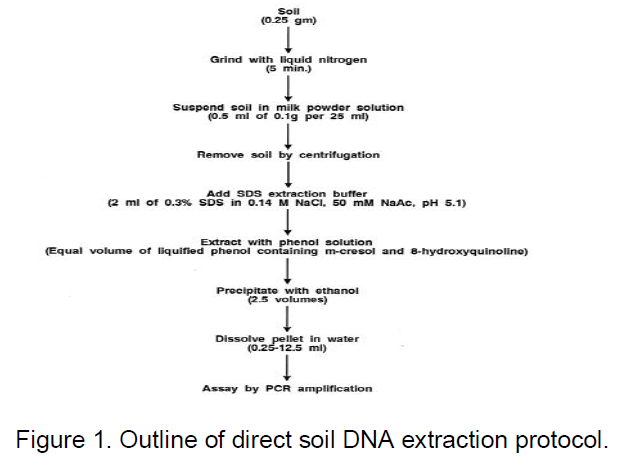
Usually the nucleic acid was extracted with SDS buffer phenol (Steele et al., 1965), a very common nucleic acid extraction procedure for biochemical or genetic analyses which also had proven to be effective with plant and fungal tissues (Nazar et al., 1991). As illustrated by the examples shown in Figure 2 (gel A), when a target organism or an internal control template was added to soil samples such basic extracts often could not be adequately amplified directly (lane a), but some signal was occasionally detected even without dilution (lane b). In many cases the signal strength could be increased significantly by further dilution of the extract (Figure 2, gel B) presumably because levels of inhibiting substances are reduced and PCR amplification remains sufficiently sensitive to permit the detection of target DNA. Because more-drastic disruption methods or conditions have previously been used for soil extracts, additional treatments also were examined; they included the use of an alkaline SDS extraction buffer, often used for DNA preparations (18); vortexing with glass beads; microwave heating or freeze-thawing to disrupt the cells (Steffan et al., 1988), (Smalla et al., 1993); and additional extraction with acetone or acetonitrile to remove substances which may interfere with PCR amplification. As illustrated in the examples shown in Figure 3, none of these treatments was found to be beneficial and most actually reduced the signal or even eliminated it entirely. For example, an alkaline SDS buffer (gel B, lane b) resulted in much higher levels of inhibition, presumably because additional inhibitors were extracted under alkaline conditions, and boiling (gel A, lane b) led to much higher losses presumably because the DNA was degraded or adsorbed. Whatever the mechanism, these methods were not helpful and were not incorporated in the standardized protocol. Because soil sample signals often remained lower than those of equivalent DNA controls even when inhibiting substances were not detected, further efforts were made to eliminate losses due to absorption or degradation. In many biochemical studies of various nucleic acids, adsorption and degradation are often minimized through the addition of nucleic acid carrier or other polyvalent polymers. In hybridization


analyses, for example, Denhardt (1966), addressed this problem by incorporating a mixture of three carrier macromolecules: 1% BSA, 1% Ficoll (Pharmacia Biotech Inc., Uppsala, Sweden), and 1% polyvinylpyrrolidone, commonly referred to as Denhardt’s solution. To evaluate the possibility that such a solution or one of the constituents might significantly reduce or eliminate losses due to degradation or adsorption, the three components, both as a complete mixture and as individual components, were added to the soil immediately prior to the extraction buffer. As illustrated in Figure 4, all were often found to significantly improve the signal strength, and when the PCR product yield was compared with the yield of control reactions containing only equivalent amounts of target DNA (gel B, lane e), the recovery was clearly high, with little loss of target DNA. In fact, a slight increase in signal strength was often observed (e.g., gel B, lane b), possibly because the carriers further stabilize the Taq DNA polymerase or enhance the reaction by some other means. Because the constituents of Denhardt’s solution are relatively expensive and not always readily available, a more common carrier was examined, namely, skim milk powder. This substance has also been reported to be effective as a carrier in reducing background signals and clearly would be inexpensive and readily available. As shown in Figure 5, with the same soil sample as used in Figure 4, the results were again very satisfactory, with 0.1 g of milk powder per 25 ml of H2O being an optimized concentration (lane b). Without milk powder virtually no signal was observed (Figure 4). Lower concentrations often resulted in a reduced signal strength (e.g., in lane a with 0.01 g of milk powder the signal is reduced by 42%), and higher concentrations resulted in streaking (e.g., lane c). Furthermore, as shown in Figure 6, when applied to typical farm soils from six diverse regions of Iran, a signal was sometimes detectable without dilution, but the signal strength was always strong when the extracts were diluted 50-fold prior to PCR amplification. As shown in Figure 7, the standard protocol (gel A) was equally successful with sand and fine gravel (lanes a, c, e, and g), but only trace or no signals were observed with clay (lanes b and f). As also shown in Figure 7, this problem could be partially overcome with the use of higher milk powder concentrations (gel B). Although quantitative analyses could remain a problem with clay samples, the target DNA was detectable. Because control template DNA was used in developing the extraction protocol, key experiments were also repeated with microsclerotia, a highly resistant storage form of V. dahlia which is commonly found in soils. As illustrated in Figure 8, the conclusions were the same for both standard and nested PCR amplification. The genomic DNA signal was relatively weak with the standard amplification protocol (gel A, lane a) but much stronger after nested PCR amplification (gel B, lane a).
As previously noted by other investigators (Haqqi et al., 1988), the application of nested PCR provides for a more dramatic level of sensitivity and permits much higher levels of dilution and diagnostics which should be able to detect almost any level of microbe activity in soil samples.



In summary, therefore, a rapid and cost-effective method to extract DNA directly from soil samples which can be utilized with PCR amplification to effectively detect specific soil organisms has been developed. Many PCR-based assays for specific organisms have already been developed and many more are certain to follow. The extraction procedure which is defined by this study should be applicable to many, if not all, of these specific assays, providing for accurate and efficient monitoring of these target organisms in soil. Extracts from samples containing large amounts of clay are less effective, but qualitative analyses are possible and the use of internal control templates should permit quantitative analyses as well.
Limitations of PCR and RT-PCR
The PCR reaction starts to generate copies of the target sequence exponentially. Only during the exponential phase of the PCR reaction is it possible to extrapolate back to determine the starting quantity of the target sequence contained in the sample. Because of inhibitors of the polymerase reaction found in the sample, reagent limitation, accumulation of pyrophosphate molecules, and self-annealing of the accumulating product, the PCR reaction eventually ceases to amplify target sequence at an exponential rate and a "plateau effect" occurs, making the end point quantification of PCR products unreliable. In summary, The discovery in 1976 of Taq polymerase -a DNA polymerase purified from the thermophilic bacterium, Thermus aquaticus, which naturally lives in hot (50 to 80°C (122 to 176 °F)) environments (Chien et al., 1976). Such as hot springs- paved the way for dramatic improvements of the PCR method. the genetic differences observed through RFLP analysis of the PCR amplified nuclear rDNA IGS region and mitochondrial SSU rRNA gene indicated intraspecific variability in V. dahliae, separating isolates from olive from those in other hosts. Further research, using a more representative set of isolates, including cross pathogenicity studies with all isolates, and full length sequencing of PCR products, will be necessary to determine if intraspecific groups continue to correlate with the plant hosts.
The authors have not declared any conflict of interest.
REFERENCES
Cheng S, Fockler C, Barnes WM, Higuchi R (1994). Effective amplification of long targets from cloned inserts and human genomic DNA". Proceed. National Acad. Sci. 91(12):5695–5699.
Crossref |
|
|
|
|
|
Chien A, Edgar DB, Trela JM (1976). "Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus, J. Bacteriol. 174 (3):1550–1557. |
|
|
Denhardt D (1966). A membrane filter technique for the detection of complementary DNA. Biochem. Biophys. Res. Commun. 23:641–646.
Crossref |
|
|
|
|
Haqqi TM, Sarkar G, David CS, Sommer SS (1988). Specific amplification with PCR of a refractory segment of genomic DNA. Nucleic Acids Res. 16:11844–11850.
Crossref |
|
|
|
|
Holmes DS, Quigley M (1981). A rapid method of preparation of bacterial plasmids. Anal. Biochem. 114:193–197.
Crossref |
|
|
|
|
Hu X, Nazar RN, Robb J (1993). Quantification of verticillium bio- mass in wilt disease development. Physiol. Mol. Plant Pathol. 42:23–36.
Crossref |
|
|
|
|
McDonnell MW, Simon MN, Studier FW (1977). Analysis of restriction fragments of T7DNA and determination of molecular weights by electrophoresis in neutral and alkaline gels. J. Mol. Biol. 110:119–124.
Crossref |
|
|
Nazar RN, Hu X, Schmidt J, Culham D, Robb EJ (1991). Potential use of PCR-amplified ribosomal intergenic sequences in the detection and differentiation of verticillium wilt pathogens. Physiol. Mol. Plant Pathol. 39:1–11.
Crossref |
|
|
|
Pillai SD, Josephenson KL, Bailey RL, Gerba C, Pepper IL (1991). Rapid method for processing soil samples for polymerase chain reaction amplification of specific gene sequences. Appl. Environ. Microbiol. 57:2283–2286. |
|
|
Radloff R, Bauer W, Vinograd J (1967). A dye-buoyant-density method for the detection and isolation of closed circular duplex DNA: the closed circular DNA in HeLa cells. Proc. Natl. Acad. Sci. USA 57:1514–1520.
Crossref |
|
|
Rogers SO, Bendich AJ (1985). Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol. Biol. 5:69–76.
Crossref |
|
|
Sharpe PA, Sugden B, Sambrook J (1973). Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose. Biochemistry 12:3055–3058.
Crossref |
|
|
|
|
Smalla K, Cresswell N, Mendonca-Hagler LC, Wolters A, van Elsas JD (1993). Rapid DNA extraction protocol from soil for polymerase chain reaction-mediated amplification. J. Appl. Bacteriol. 74:78–85.
Crossref |
|
|
|
|
Steele WS, Okamura N, Busch H (1965). Effects of thioacetamide on the composition and biosynthesis of nucleolar and nuclear ribonucleic acid in rat liver. J. Biol. Chem. 240:1742–1749.
PMid:14285518 |
|
|
Steffan RJ, Atlas RM (1991). Polymerase chain reaction: Application in environmental microbiology. Annu. Rev. Microbiol. 45:137–161.
Crossref
|
|
|
|
|
|
Steffan RJ, Goksoyr J, Bej AR, Atlas RM (1988). Recovery of DNA from soils and sediments. Appl. Environ. Microbiol. 54:2908–2915. |
|
|
|
Tuite J (1969). Plant pathological methods: fungi and bacteria. Burgess Publishing Co., Minneapolis, Minn. |