ABSTRACT
Iris yellow spot virus (IYSV) is an important pathogen of Allium species worldwide. It has a tripartite genome consisting of the large (L), medium (M) and small (S) RNA segments. Despite its worldwide distribution, very few complete gene and genome sequences are available in public databases. The aim of this study was to obtain full gene sequences of a garlic-infecting IYSV isolate by next-generation sequencing (NGS) for understanding its evolution. Total RNA was extracted from an IYSV-positive garlic leaf and sequenced on the Illumina HiSeq platform using paired-end chemistry 125 × 125 bp reads. The quality of raw reads was assessed using FastQC software before trimming with Trimmomatic version 0.36. The resultant paired-end sequences were used for both de novo and reference-based genome assembly. The resultant consensus gene sequences were analyzed using SIAS (for sequence identity and composition), ExPASy (for protein molecular weight) and ORF Finder (for open reading frame identification). Three full gene sequences, that is, nucleocapsid (N), nonstructural protein (NSs) and movement protein (NSm) were recovered. The N gene did not display any distinct clustering patterns based on geographical locations and was most identical to an onion-infecting isolate from Serbia (Accession KT272878). The NSs and NSm genes clustered closely with homologous sequences of IYSV isolates that were retrieved from GenBank and EMBL. This study lays the foundation for complete genome studies of IYSV in Zimbabwe.
Key words: Allium species, emerging pathogen, reverse transcription polymerase chain reaction (RT-PCR), serology, tospovirus.
Iris yellow spot virus, IYSV, is an important emerging pathogen of Allium species worldwide responsible for causing significant yield and quality losses in alliaceous and ornamental crops (Bag et al., 2015). It was first isolated and characterized in iris (Iris holandica) in The Netherlands (Cortês et al., 1998) and has now been confirmed to be present in over 40 countries worldwide (CABI, 2018). In Zimbabwe, IYSV was first detected infecting onions (Allium cepa) in 2014 (Karavina et al., 2016) and was subsequently reported infecting garlic (Allium sativum), leeks (Allium ampeloprasum) and shallots (A. cepa var. aggregatum) (Karavina and Gubba, 2017).
IYSV belongs to the genus virus in the family Tospoviridae. Like other tospoviruses, IYSV has quasi-spherical enveloped particles that are 80 to 120 nm in diameter (Pappu et al., 2009). It has a tripartite single-stranded RNA genome consisting of the large (L), medium (M) and small (S) segments. The L RNA encodes the RNA-dependent RNA polymerase (RdRp) in the viral complementary sense (Bag et al., 2010). The M RNA is ambisense and has two open reading frames (ORFs). The viral sense strand of the M RNA encodes the non-structural movement (NSm) protein, while the anti-viral sense strand encodes the Gn/Gc glycoproteins. The NSm protein is responsible for virus particle movement during systemic infection, while the two glycoproteins serve as virus attachment proteins (Bandla et al., 1998). The S RNA is also ambisense and encodes the non-structural (NSs) protein in the viral sense strand and the nucleocapsid (N) protein in the antiviral sense strand. The NSs protein is involved in suppressing RNA silencing, while the N protein encapsidates the RNA segments (Bag et al., 2015). In addition to these genes, both M and S RNAs contain intergenic regions (IGRs) capable of forming stable hairpin structures (King et al., 2012). IYSV is currently known to be transmitted by two thrips species, Thrips tabaci and Frankliniella fusca in a persistent and propagative mode (Srinivasan et al., 2012).
IYSV identification and characterization is carried out by employing biological, serological, morphological and molecular techniques (Bag et al., 2015). Biological characterization, also known as host indexing, involves the inoculation of indicator plant species that produce typical symptoms. However, it normally takes several days for symptoms to develop on inoculated hosts (Bag et al., 2012). Serological detection can lead to ambiguous results, especially amongst closely related viruses like IYSV and Tomato yellow ring virus (TYRV). Also, antibodies to IYSV must be raised if serological detection is to be successful. Morphological identification involves the use of electron microscopy, a technique that is technically challenging and not affordable in most research and academic institutions in developing countries. Being RNA viruses, reverse transcription-polymerase chain reaction (RT-PCR) can also be employed for IYSV identification (Walsh et al., 2001). However, primers specific for IYSV detection must be available/designed.
Next-Generation Sequencing (NGS) provides an unbiased, quick, cost- and labour-effective characterization procedure for plant viruses (Kreuze et al., 2009). Two assembly methods, namely, de novo assembly and reference-based mapping are used to recover the virus genome from the sequenced data. With NGS, full viral genomes can be recovered at once (Adams et al., 2009).
In public databases like NCBI and EMBL, except for the N gene, there are no more than five complete IYSV gene sequences available. This has negatively impacted evolutionary studies of this pathogen. In this study, NGS was used to characterize three full genes of a garlic-infecting IYSV isolate from Zimbabwe.
Sources of materials used
IYSV was isolated from garlic plants collected at a horticultural farm in Chegutu, Zimbabwe, in 2015. Garlic plants that displayed necrotic, irregularly-shaped and grey-to-bleached lesions typical of IYSV infection were targeted during sample collection. Young leaves from symptomatic plants were collected in RNAlater® solution (Thermo Fisher Scientific, USA) and transported to the University of KwaZulu-Natal (South Africa) for pathogen characterization.
Serological detection
Eight leaf samples were tested for IYSV in duplicate wells using a commercial kit supplied by Loewe® Biochemica GmbH (Sauerlach, Germany) following the manufacturer’s instructions. Briefly, microtiter plates were coated with IYSV-specific antibodies. About 0.5 g of garlic leaf tissue showing disease symptoms were excised and ground in liquid nitrogen in a pestle and mortar. Macerated tissues were mixed with Conjugate Buffer at 1:20 dilution, and 0.2 ml mixture added to each microtiter well before overnight incubation. After four washes, an enzyme-labelled antibody-AP-conjugate was applied to the plate wells. In the final step, 0.2 ml of the Substrate Buffer Solution containing the dissolved substrate was applied to the microtiter plate. After 2 h of incubation, the reaction was visually assessed for yellow color development which is characteristic of a positive test.
RNA extraction and RT-PCR
Total RNA was extracted from the garlic leaves using the Quick RNA MiniPrep Kit (Zymo Research, USA) according to manufacturer’s instructions. RT-PCR was performed using the RevertAid RT Reverse Transcription Kit (Thermo Fischer Scientific, USA) targeting the N gene. The resultant cDNA was used for PCR amplification using the KAPA2G Fast Hot Start ReadyMix Kit (KAPA Biosystems, USA) and N gene-specific primers (IYSV_2F: 5'-GGCGGTCCTCTCATCTTACTG-3' and IYSV-NCP2_R: 5'-GAAGTTCCAGGAGTGCATTTAGTC-3') (Lee et al., 2011) under the following cycling conditions: initial denaturation at 95°C for 2 min; 35 cycles of 94°C for 15 s (denaturation step), 57°C for 15 s (annealing step), and 72°C for 15 s (extension step), followed by a 5-min incubation period at 72°C. PCR products were analyzed by
1.5% agarose gel electrophoresis. The amplicons were excised, purified using a ZymocleanTM Gel DNA Recovery Kit (Zymo Research, USA) and directly sequenced in both directions at Inqaba Biotechnical Industries (Pty) Ltd (Pretoria, South Africa). Sequences generated were blasted into MEGA 6.06 program.
Sample preparation, sequencing and data analysis for NGS
A sample that was IYSV-positive by both DAS-ELISA and RT-PCR was randomly selected and RNA extracted for NGS at the Agricultural Research Council’s Biotechnology Platform (ARC-BTP) (Pretoria, South Africa). RNA quality and quantity were assessed using a nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, USA). Total RNA samples were pre-treated with Ribo-Zero (NEB, UK) prior to library preparation.
NGS was performed on the Illumina HiSeq platform using paired`-end chemistry 125 × 125 bp reads. Subsequent sample demultiplexing was done using the CASAVA pipeline software (Illumina, USA). Read lengths of less than 25 nucleotides were trimmed and pair-end sequence libraries were generated. FastQC version 0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/ fastqc/ ) was used to assess quality of the raw reads generated by NGS before and after trimming with the Trimmomatic v 0.36 with the following settings:
(ILLUMINACLIP: TrueSeq3-PE-2.fa:2:30:9:1:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15:MINLEN:36) (Bolger et al., 2014).
The pair-end sequences were subsequently used for both de novo assembly and reference-based mapping. De novo assembly was performed using the SPAdes Genome Assembler version 3.10.1 with the parameters: careful mode; only assembler; –k 21, 33, 55, 77, 99 (Bankevich et al., 2012), while referenced-based genome mapping was done using the BBMap Short Read Aligner version 37.28 (Bushnell, 2014) using IYSV genomes as references. All contigs generated by the de novo assembly method were subjected to BLAST using ncbi-blast 2.6.0+ (www.ncbi.nih.gov).
IYSV sequence analysis and phylogeny
All contigs that matched IYSV genomes were selected and aligned using the Clustal W embedded in MEGA 6.06 software (Tamura et al., 2013) to generate the consensus sequences of the N, NSs and NSm genes. The open reading frames (ORFs) on the IYSV genes were identified using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder). Molecular weight (Mw) of proteins was determined using the online ExPASy bioinformatics tool (Gasteiger et al., 2005). Phylogenetic trees of the nucleotide sequences of the three IYSV ORFs were inferred by Maximum Likelihood method based on the best evolutionary models as determined by MEGA 6.06. The trees were rooted using the TSWV sequence as an outgroup. Bootstrap analyses were conducted using 1000 replicates. Details of isolates used in the phylogenetic analysis are shown in Table 3. Nucleotide and amino acid sequence compositions and sequence identities were calculated using SIAS program (www.imed.ucm.es/Tools/sias.html).
Serology and RT-PCR analyses
Of the eight samples that were tested, six were positive for IYSV by both DAS-ELISA and RT-PCR. All IYSV- positive samples turned yellow within 2 h of incubation after the final wash step. The 236-bp bands were visualized from samples that were IYSV-positive after electrophoresis on 1.5% agarose gel stained with SYBR Safe Gel stain (Life Technologies, USA).
RNA quality assessment and NGS data analysis
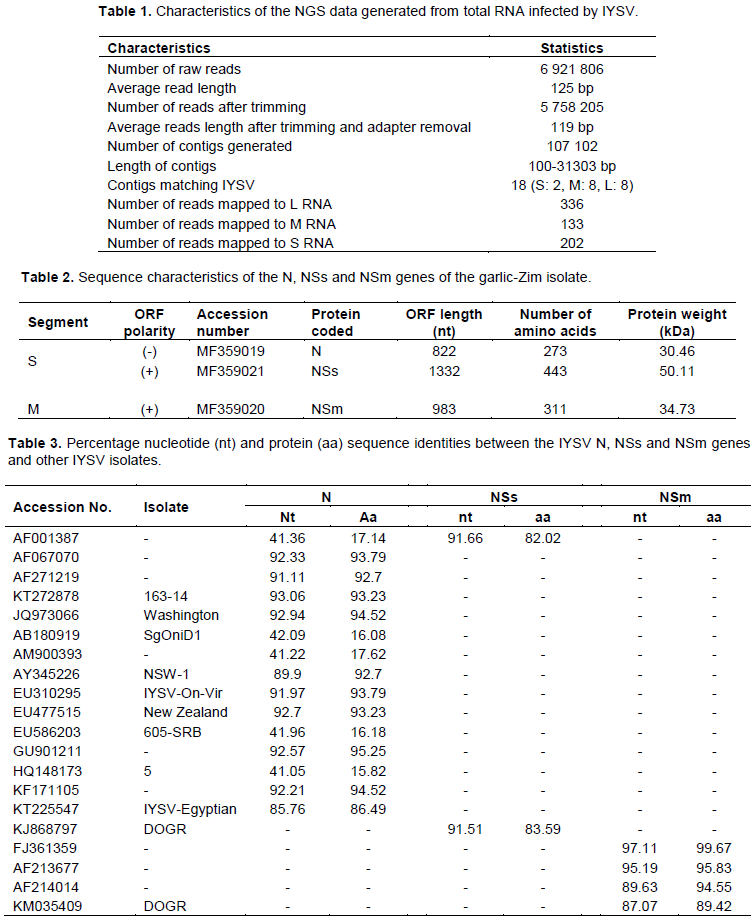
The concentration of the RNA sample that was sent for NGS was 194.38 ng/µL, with an absorbance A260/A280 ratio of 1.95. The size of the NGS data generated was 3.3 GB and consisted of 6 921 806 raw reads. The average reads length after trimming was 119 bp (Table 1). A total of 107 102 contigs were generated by de novo analysis. Of these, 18 matched to the L (2), M (8) and S (8) RNA segments of the IYSV genome. From the reference-based mapping, a total of 671 reads were mapped to the IYSV genome (Table 1). Although the results obtained using both de novo assembly and reference-based mapping methods were consistent, full IYSV genome segments of the garlic Zimbabwe (garlic-Zim) isolate were not recovered. However, three genes (N and NSs found on the S RNA segment and the NSm present on the M RNA segment) were found to be complete after visual inspection and analysis on the ORF Finder. Consequently, these genes were considered for phylogenetic analyses. The nucleotide sequences of these genes were deposited in GenBank under the accession numbers shown in Table 2.
Sequence characteristics and phylogenetic analyses of the N, NSs and NSm genes were from the IYSV garlic-Zim isolate The N gene of the IYSV garlic-Zim isolate was 822 nt long and coded for a protein with a molecular weight of 30.46 kDa. The NSs and NSm proteins had molecular weights of 50.11 and 34.73 kDa, respectively (Table 2).
The N gene of the garlic-Zim isolate had a sequence identity of 93.06% to the onion-infecting Serbian isolate (KT272878) at the nucleotide level, while at the protein level; it was most identical (95.25%) to the onion-infecting Sri Lankan isolate (GU901211). It shared the lowest nucleotide and protein sequence identity with the Iranian isolate (HQ148173). The NSs gene sequence of the garlic-Zim isolate was 91.66% identical to The Netherlands isolate (AF001387) at the nucleotide level. As for the NSm gene, it was most identical to the USA isolate (FJ361359) at both nucleotide and amino acid levels (Table 3).
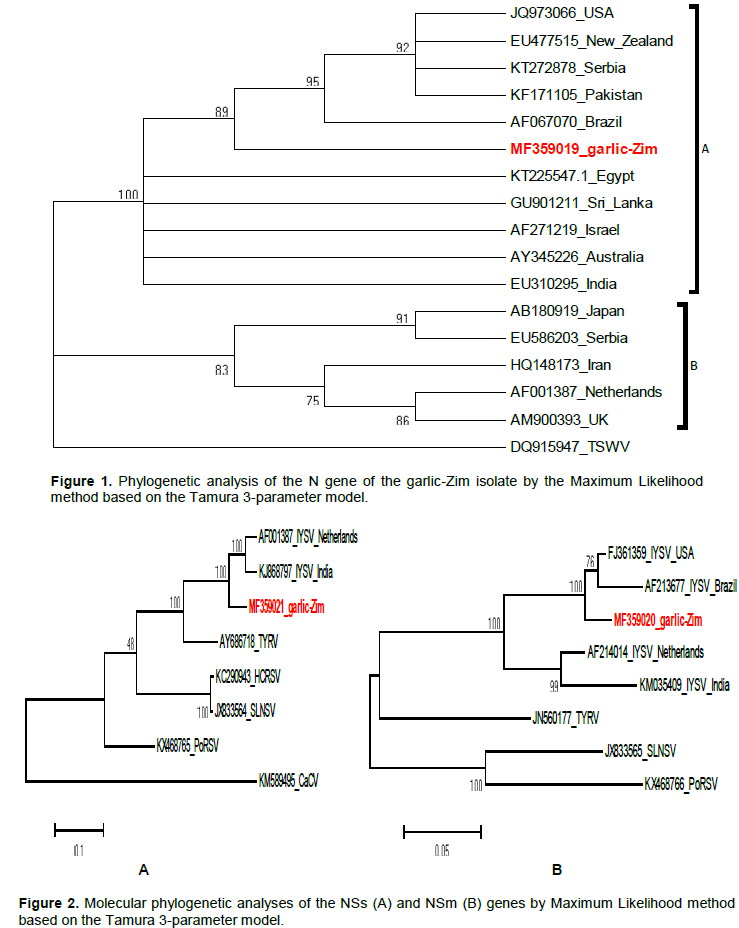
Phylogenetic analysis of the N genes produced two distinct clusters (A and B; Figure 1). The N gene of the IYSV garlic-Zim isolate was in cluster A along with homologous sequences of isolates from Australia, Brazil, Egypt, India, Israel and Sri Lanka. Cluster B composed of N sequences of isolates from Iran, Japan, Serbia, The Netherlands and The UK (Figure 1).
The NSs gene of the garlic-Zim isolate clustered with the homologous sequences of isolates from India and
The Netherlands (Figure 2A). As for the NSm gene, it also clustered with homologous sequences of IYSV isolates from Brazil (AF213677) and The USA (FJ361359) (Figure 2B).
The genomic organization of the garlic-Zim isolate is typical of tospoviruses in being tripartite (Pappu et al., 2009). Though no full genomic segments were recovered, NGS enabled the simultaneous recovery of the two full genes on the S RNA segment and one gene on the M segment. This is not possible with Sanger sequencing. This study lays the foundation for future studies on the full genome of IYSV in Zimbabwe.
Despite the global importance of IYSV, only a few full genome and gene sequences have been characterized (Gawande et al., 2015). This greatly compromises studies to understand pathogen evolution and management. The current study is first in Africa to characterize more than one full gene sequence of the same IYSV isolate. The only other full IYSV gene sequences from Africa deposited in public databases are from Egypt (Accessions KT225547.1 and KC161369.1).
Phylogenetic analysis of the N gene showed no specific clustering patterns based on geographical locations. This could suggest the possibilities of long-distance migration, recombination and reassortment events in IYSV. Such events are highly prevalent in tospoviruses (Margaria et al., 2015; Zhang et al., 2016). Being a pathogen of some internationally-traded ornamental plants (Bag et al., 2015; CABI, 2018), it is possible that IYSV and its vectors have been unintentionally distributed worldwide in live plant shipments. Also, the smuggling of live host plants across borders could have contributed to pathogen’s worldwide distribution. The S segment is known to be substantially more prone to recombination than the M and L segments (Gawande et al., 2015). For the occurrence of either recombination or reassortment to be confirmed in the Gar-Zim isolate, full genomic segments must be recovered and analyzed.
In addition to DAS-ELSIA and serology, NGS was employed in further confirming the occurrence of IYSV in Zimbabwe. Knowledge of IYSV presence is crucial in epidemiological studies towards developing effective disease control strategies. When compared to serology and RT-PCR, NGS is a more rapid procedure and it also produces nucleic acid sequences for substantial parts of the viral genome. Another major advantage of NGS over DAS-ELISA and RT-PCR is that the latter approaches require reagents designed exclusively to detect their viral target and any variation in the virus genome may cause the assay to fail. NGS is non-targeted and requires no prior knowledge of the target. Therefore, it can detect existing strains, new variants and even new strains (Adams et al., 2009).
To maximize the chances of detecting IYSV in the sequenced data, two different methods (de novo assembly and reference-based mapping) were employed. De novo assembly recreates the original genome sequence through overlapping reads while reference-based mapping requires a previously assembled genome to be used as a reference. A major advantage of de novo assembly over reference-based mapping is that it gives the virome of the host(s) studied. It also detects other viruses not targeted by the study (Martin and Wang, 2011). In this study, Garlic common latent virus, Garlic virus B, Garlic virus C and Shallot virus X were detected (data not shown). There were no significant differences in the IYSV genes that were recovered by both de novo and reference-based mapping.
The fact that viruses other than IYSV were detected in the sample that was sent for NGS shows that mixed and multiple infections are common in nature. This implies that symptom expression cannot be conclusively relied upon for disease diagnosis.
The characterized IYSV genes of the garlic-Zim isolate are the foundation for future full genome studies of this important pathogen. Knowledge of the full genome is critical in understanding the evolutionary patterns of IYSV. Pathogen genomic information is also important in developing IYSV disease management strategies.
The authors have not declared any conflict of interests.
The authors would like to thank The Kellogg Foundation Southern African Scholarship Program for financial support.
REFERENCES
|
Adams IP, Glover RH, Monger WA, Mumford R, Jackeviciene E, Navalinskiene M, Samuitiene M, Boonham N (2009). Next-generation sequencing and metagenomics analysis: a universal diagnostic tool in plant virology. Molecular Plant Pathology 10(4):537-545.
Crossref
|
|
|
|
Bag S, Druffel KL, Pappu HR (2010). Structure and genome organization of the large RNA of Iris yellow spot virus (genus Tospovirus, family Bunyaviridae). Archives of Virology 155:275-279.
Crossref
|
|
|
|
|
Bag S, Schwartz HF, Cramer CS, Harvey MJ, Pappu HR (2015). Iris yellow spot virus (Tospovirus: Bunyaviridae): from obscurity to research priority. Molecular Plant Pathology 16(3):224-237.
Crossref
|
|
|
|
|
Bag S, Schwartz HF, Pappu HR (2012). Identification and characterization of biologically distinct isolates of Iris yellow spot virus (genus Tospovirus, family Bunyaviridae), a serious pathogen of onion. European Journal of Plant Pathology 134:97-104.
Crossref
|
|
|
|
|
Bandla MD, Campbell LR, Ullman DE, Sherwood JL (1998). Interaction of Tomato spotted wilt tospovirus (TSWV) glycoproteins with a thrips midgut protein, a potential cellular receptor for TSWV. Phytopathology 88:98-104.
Crossref
|
|
|
|
|
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19:455-477.
Crossref
|
|
|
|
|
Bolger AM, Lohse M, Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114-2120.
Crossref
|
|
|
|
|
Bushnell B (2014). BBMap short read aligner.
View. Accessed on June 11, 2018.
|
|
|
|
|
CABI (2018). Iris yellow spot virus datasheet 28848.
View. Accessed on July 7, 2018.
|
|
|
|
|
Cortês I, Livieratos IC, Derks A, Kormelink R (1998). Molecular and serological characterization of Iris yellow spot virus, a new and distinct Tospovirus species. Phytopathology 88:1276-1282.
Crossref
|
|
|
|
|
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005). Protein identification and analysis tools on the ExPASy Server. In: The Proteomics Protocols Handbook. J.M. Walker (eds). Humana Press, USA.
Crossref
|
|
|
|
|
Gawande SJ, Gurav VS, Martin DP, Asokan R, Gopal J (2015). Sequence analysis of Indian Iris yellow spot virus ambisense genomic segments: evidence of interspecies RNA recombination. Archives of Virology 160:1285-1289.
Crossref
|
|
|
|
|
Karavina C, Gubba A (2017). Iris yellow spot virus in Zimbabwe: Incidence, severity and characterization of Allium-infecting isolates. Crop Protection 94:69-76.
Crossref
|
|
|
|
|
Karavina C, Ibaba JD, Gubba A (2016). First report of Iris yellow spot virus infecting onion in Zimbabwe. Plant Disease 100(1):235.
Crossref
|
|
|
|
|
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (2012). Virus taxonomy- classification and nomenclature of viruses. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, Amsterdam, The Netherlands pp. 725-741.
|
|
|
|
|
Kreuze JF, Perez A, Untiveros M, Quispe D, Fuentes S, Barker I, Simon R (2009). Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a genetic method for diagnosis discovery and sequencing of viruses. Virology 388:1-7.
Crossref
|
|
|
|
|
Lee J-S, Cho WK, Choi H-S, Kim K-H (2011). RT-PCR detection of five quarantine plant RNA viruses belonging to poty- and tospo-viruses. Plant Patholology Journal 27(3):291-296.
Crossref
|
|
|
|
|
Margaria P, Miozzi L, Ciuffo M, Pappu H, Turina M (2015). The first complete genome sequences of two distinct European Tomato spotted wilt virus isolates. Archives of Virology 160:591-595.
Crossref
|
|
|
|
|
Martin JA, Wang Z (2011). Next-generation transcriptome assembly. Nature Reviews Genetics 12:671-682.
Crossref
|
|
|
|
|
Pappu HR, Jones RAC, Jain RK (2009). Global status of tospovirus epidemics in diverse cropping systems: Successes achieved and challenges ahead. Virus Research 141:219-236.
Crossref
|
|
|
|
|
Srinivasan R, Sundaraj S, Pappu HR, Diffie S, Riley DG (2012). Transmission of Iris yellow spot virus by Frankliniella occidentalis and Thrips tabaci (Thysanoptera: Thripidae). Journal of Economic Entomology 105:40-47.
Crossref
|
|
|
|
|
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30:2725-2729.
Crossref
|
|
|
|
|
Walsh K, North J, Barker I, Boonham N (2001). Detection of different strains of Potato virus Y and their mixed infections using competitive fluorescent RT-PCR. Journal of Virological Methods 91:167-173.
Crossref
|
|
|
|
|
Zhang Z, Wang D, Yu C, Wang Z, Dong J, Shi K, Yuan X (2016). Identification of three new isolates of Tomato spotted wilt virus from different hosts in China: molecular diversity, phylogenetic and recombination analyses. Virology Journal 13:8.
Crossref
|
|