ABSTRACT
Malaria remains the global public health problem due to the reemergence of drug resistance. There is an urgent need for development of new antimalarial candidates which are effective against resistant malaria parasite. This systematic review evaluates the published research studies that applied in silico modeling during the discovery process of antimalarial drugs. Literature searches were conducted using PubMed, EBSCO, EMBASE, and Web of Science to identify the relevant articles using the search terms “Malaria” “In silico model”, “Computer-based drug design”, “Antimalarial drug”, and “Drug discovery”. Only the articles published in English between 2008 and May 2015 were included in the analysis. A total of 17 relevant articles met the search criteria. Most articles are studies specific to Plasmodium falciparum targets; 3 and 1 articles, respectively involve target for P. vivax and liver stage of Plasmodium. Both structure-based and ligand-based approaches were applied to obtain lead antimalarial candidates. Two articles also assessed absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. Confirmation of activity of the candidate leads by in vitro and/or in vivo assays were reported in some studies. Homology modelling, molecular docking, 2D- or 3D-QSAR and pharmacophore modeling are commonly applied methods. One study used de novo synthesis for lead identification and one study applied phylogenetic analysis for target identification/validation.
Key words: Plasmodium, malaria, antimalarial drug, drug discovery, in silico modeling, computer-based drug design, systematic review.
Malaria is one of the most important global infectious diseases affecting hundreds of millions of people each year and is the main cause of socio-economic loss in developing countries (WHO, 2015). The major problem in malaria treatment and control is the emergence and spread of multidrug resistance Plasmodium falciparum including artemisinin drugs. It is therefore an urgent need to search for alternative effective drug candidates. Nevertheless, the process of drug discovery and development is both time-consuming (approximately 10-15 years) and resource-consuming (approximately $1.5 billion per successful drug) (Ren et al., 2016). Rational drug design is an intensive process of finding new medications based on knowledge of a biological target. Drug design involves the design of small molecules that are complementary in shape and charge to the biomolecular target with which they interact. Advances in genomics and computational methods over the past decades present new challenges and opportunities in drug discovery and development process. The convergence of these technological trends has been a great benefit to computational science and informatics. Information on gene expression, drug-target interactions (DTI), including protein networks, which are rapidly accumulating are becoming increasingly accessible and standardized (Margolis et al., 2014).
Computational-based or in silico approach can facilitate the discovery process for antimalarial drugs through utilizing a number of available databases of chemical compounds and Plasmodium proteins. The costs are minimal, humans are rarely at risk, and biosafety facilities are not required. Most drugs in development fail during clinical trials due to poor pharmacokinetics properties and toxicity. These properties such as absorption, distribution, metabolism, excretion including toxicity (ADMET) play an important role in drug discovery and development. ADMET prediction can help eliminate compounds with unfavorable drug ability properties. In silico modeling can be applied for prediction of either pharmacodynamics (targets of drug action and toxicity) or pharmacokinetic properties of the candidate molecules (Mendez et al., 2016). Both properties involve interactions with multiple biological systems (Hodos et al., 2016; Lounnas et al., 2013; Peltenburg et al., 2013). Two approaches have been successfully applied for rational drug design, that is ligand-based and structure-based drug design (Lounnas et al., 2013; March-Vila et al., 2017). Ligand-based drug design or indirect drug design relies on knowledge of other molecules that bind to the biological target of interest.
These molecules are used to drive a “pharmacophore” model that defines the minimum necessary characteristics a molecule must possess in order to bind to the target. A model of the biological target may also be used to design new molecular entities that interact with the target. The structure-based drug design or direct drug design relies on knowledge of the 3-dimensional structure of the biological target obtained through X-ray crystallography or NMR spectroscopy. 3D-QSAR refers to the application of force field calculations requiring three-dimensional structures, for example based on protein crystallography or molecule superimposition. Using the structure of the biological target, candidate drugs that are predicted to bind with high affinity and selectivity to the target may be designed using interactive graphics and the intuition of a medicinal chemist or various automated computational procedures to suggest new drug candidates (Lounnas et al., 2013; March-Vila et al., 2017). This systematic review focused on the analysis of research articles which applied computational approaches for antimalarial drug discovery. The information is useful for further development of effective antimalarial drugs
Literature searches
The following electronic databases were searched for research articles published during 2013 and 2017: PubMed, EBSCO, EMBASE, and Web of Science and Google Scholar. Thesaurus and free-text searches were also performed across each database to combine the terms “Malaria” “In silico model”, “Computer-based drug design”, “Antimalarial drug”, and “Drug discovery”. Duplicate articles were recorded and excluded from the analysis.
Eligibility criteria
Research articles published in English between 2008 and 2017 on the application of in silico (computer-based) modeling approach for discovery of antimalarial targets and lead candidates were included in the analysis. The number of papers referenced was likely an under-representation of the overall body of literature on in silico modeling due to the strict search criteria and the search terms used.
Study selection process
The searched articles from all databases were downloaded into Endnote and merged to remove duplicates. The titles and abstracts of all search articles were initially screened for potential relevance. The articles for which there was uncertainty about relevance were retained and the full texts were further reviewed. Articles that did not meet the inclusion criteria were excluded. Full-texts of all articles that met the eligibility criteria were downloaded into Endnote database.
Data extraction
The following information were extracted from each of the included article: the names of the authors, article title, journal and volume/issue published, publication year, software packages and algorithms applied, approach applied (structure-based design, ligand-based design), computational methods (homology modeling, molecular docking, pharmacophore modeling, 2- or 3-D quantitative structure activity relationship: QSAR), experimental methods (in vitro, in vivo assays), targets, hit or lead compounds, sets of database used, and server used.
Study selection
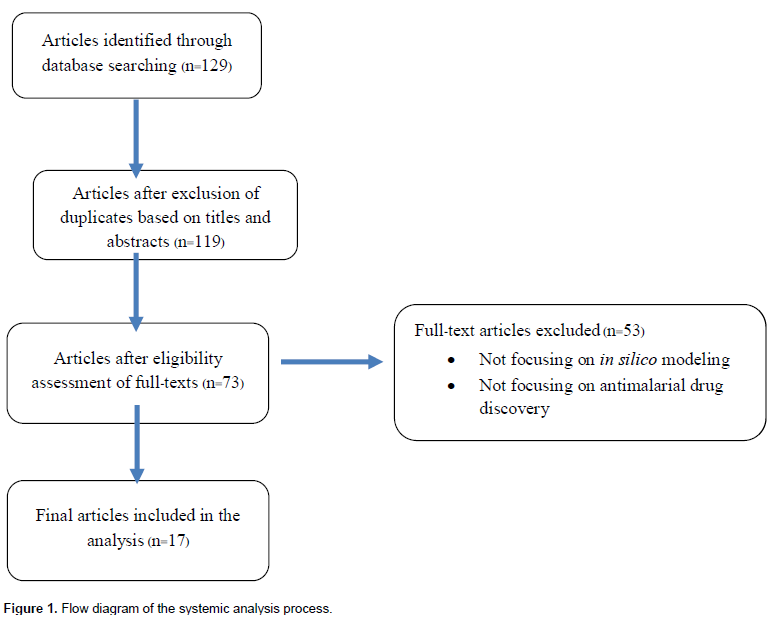
A comprehensive search of databases provided 129 potentially related articles based on the search terms described above which were published since 2008 (Figure 1). Of these articles, 10 duplicate articles were initially excluded based on the screening of titles and abstracts. Forty-six out of the 119 full-text articles were excluded from the analysis based on the eligibility criteria. Fifty-three articles were further excluded due to unrelated topics and finally 17 articles were included in the analysis.
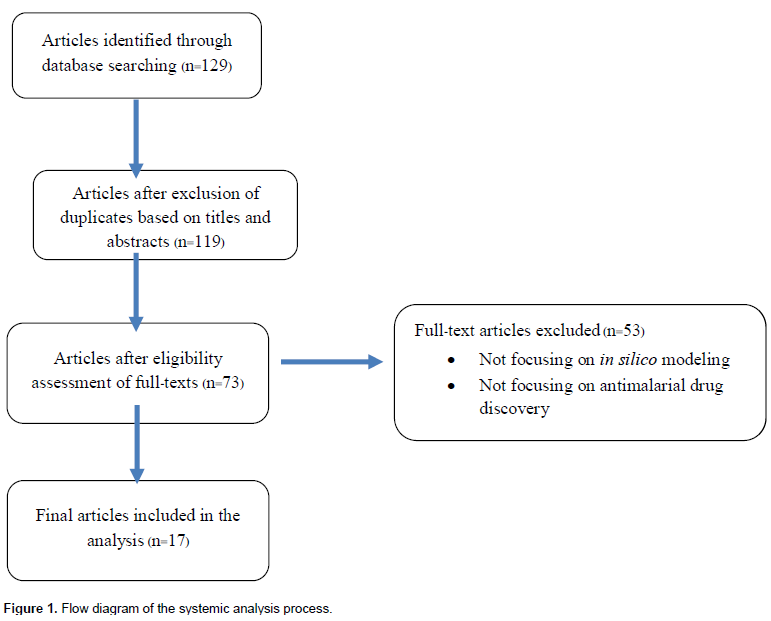
Study characteristics
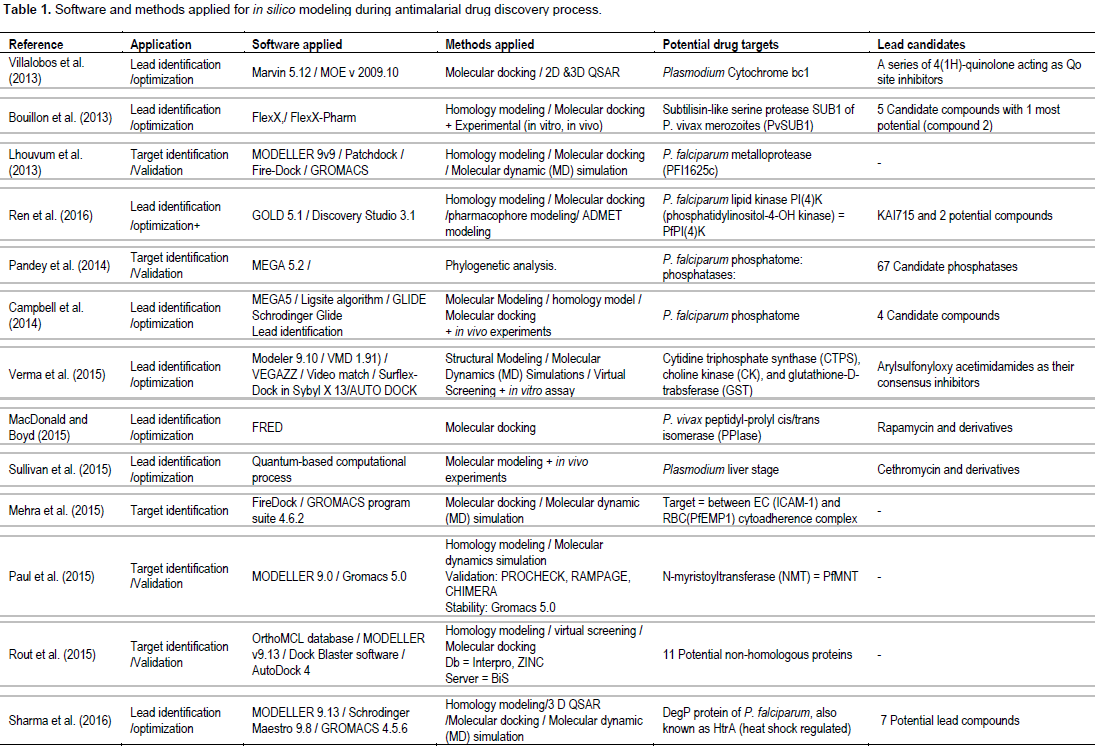
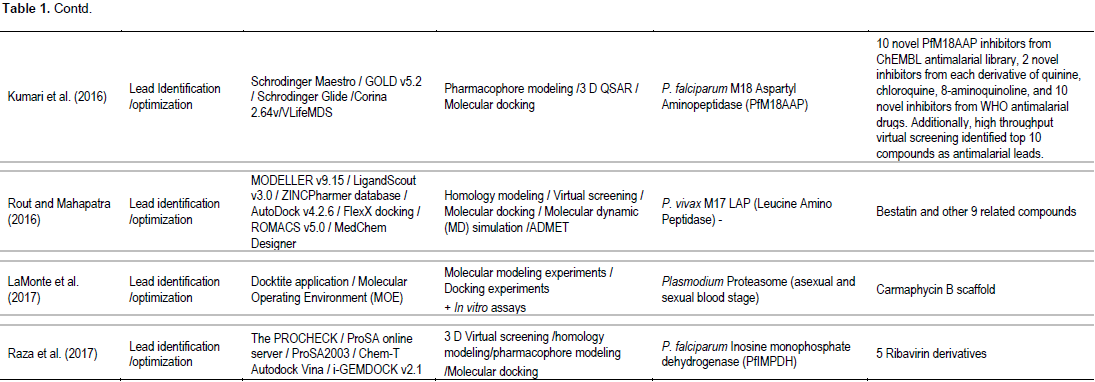
Table 1 summarizes the approaches and methodologies employed including potential targets and lead candidates from the 17 research articles included in the current systematic review. The published in silico models were mostly applied for lead identification/optimization (13 articles) of the known Plasmodium targets (13 targets). Most articles are specific for P. falciparum targets (Campbell et al., 2014; Kumari et al., 2016; LaMonte et al., 2017; Lhouvum et al., 2013; MacDonald and Boyd, 2015; Raza et al., 2017; Ren et al., 2016; Sharma et al., 2016); 3 articles involved target for P. vivax (MacDonald et al., 2015; Rout et al., 2016; Bouillon et al., 2013) and 1 article involved target for liver stage of Plasmodium (Sullivan et al., 2015). Five articles were applied in silico modeling for target identification/validation (Lhouvum et al., 2013; Mehra et al., 2015; Paul et al., 2015; Rout et al., 2015; Pandey et al., 2014). Both structure-based (Bouillon et al., 2013; LaMonte et al., 2017; MacDonald and Boyd, 2015; Raza et al., 2017; Ren et al., 2016; Rout and Mahapatra, 2016; Sullivan et al., 2015) and ligand-based (Villalobos et al., 2013; Kumari et al., 2016; Ren et al., 2016; Sharma et al., 2016) approaches were applied to obtain lead antimalarial candidates. Two articles also assessed ADMET properties (Ren et al., 2016; Rout and Mahapatra, 2016). Confirmation of activity of the candidate leads by in vitro and/or in vivo assays were reported in some studies (Bouillon et al., 2013;
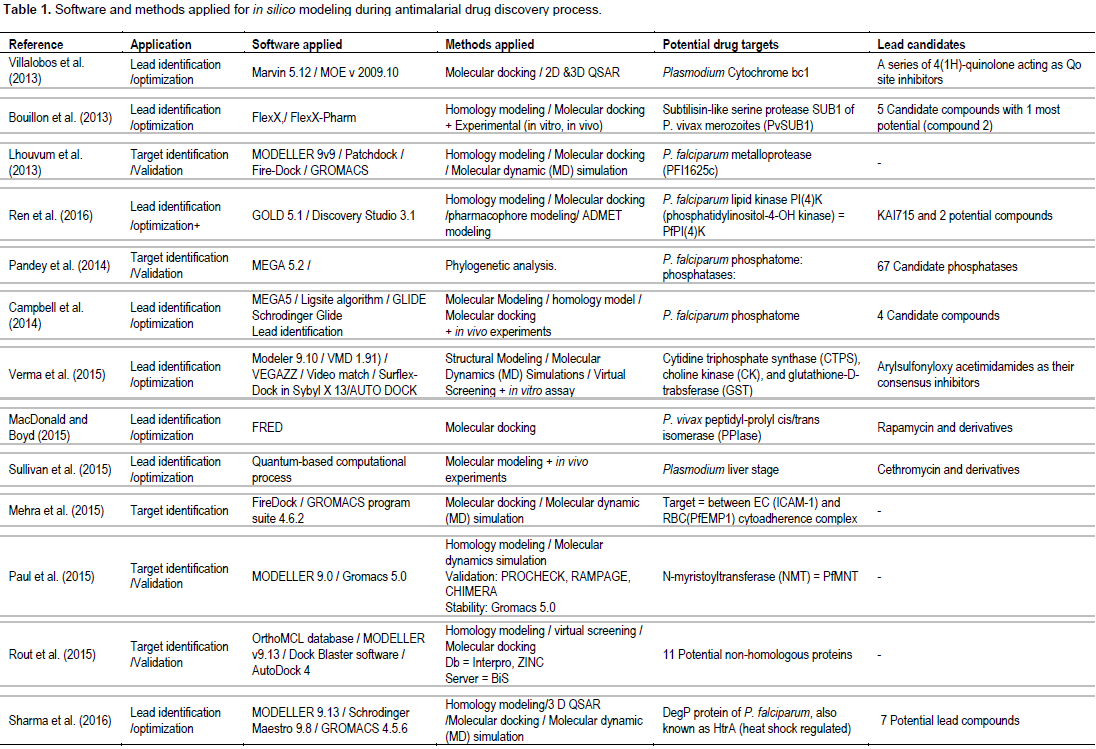
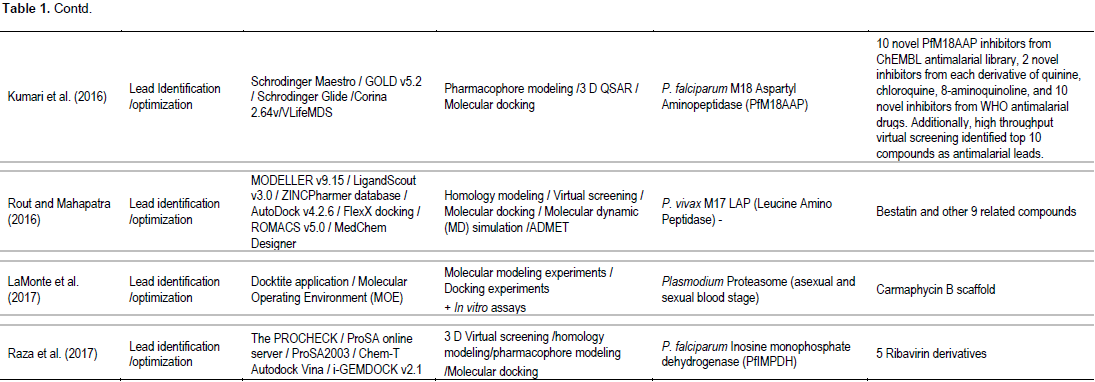
Campbell et al., 2014; LaMonte et al., 2017; Sullivan et al., 2015). Homology modelling, molecular docking, 2D- or 3D-QSAR and pharmacophore modeling were commonly applied methods. One study used de novo synthesis for lead identification (Rout and Mahapatra, 2016) and one study applied phylogenetic analysis for target identification/ validation (Pandey et al., 2014). The articles included in the analysis relied on diverse sets of software tools and web servers. Several software tools, servers, and frameworks were explicitly applied (Maier and Labute, 2014; Webb and Sali, 2014). The most commonly used software were MODELLER and GROMACS. In general, authors reported computational tools directly employed in the analysis (for example, bioinformatic web servers, molecular dynamics suites, and visualization programs) and did not explicitly report indirectly employed computational tools (for example, scripting languages, server operating systems, and clusters). Lead identification/optimization involved identification of potent antimalarial candidate molecules which are inhibitors of known specific Plasmodium targets (13 targets) encoded by the malaria parasite genome. These included: inosine monophosphate dehydrogenase (IMPDH) (Raza et al., 2017), proteasome (LaMonte et al., 2017), lipid kinase PI(4)K (phosphatidylinositol-4-OH kinase) (Ren et al., 2016), heat shock regulated A (HrA or DegP) (Sharma et al., 2016), P. falciparum M18 (Kumari et al., 2016), P. falciparum aspartyl aminopeptidase (PfM18AAP) (Kumari et al., 2016), leucine amino peptidase (M17 LAP) (Rout and Mahapatra, 2016), P. falciparum glycogen synthase kinase-3 (PfGSK-3), P. falciparum CTP synthetase (PfCTPS), choline kinase (PfCK), and glutathione S-transferase (PfGST) (Verma et al., 2015), Plasmodium peptidyl-prolyl cis/trans isomerase (MacDonald and Boyd, 2015), Plasmodium phosphatases, cytochrome bc1 (Jimenez et al., 2013), and P. vivax subtilin-like serine protease (PvSUB1) (Bouillon et al., 2013). Application of the modeling for target identification/validation included N-myristoyltransferase (NMT) (Paul et al., 2015), Plasmodium phosphatases (Campbell et al., 2014; Pandey et al., 2014), and PFI1625c metalloprotease (Lhouvum et al., 2013).
One articles involving ICAM-1 and PfEMP1 cytoadherence complex applied the computer-based modeling for both target identification/validation and lead identification/optimization (Mehra et al., 2015). Various commercial and public databases of proteins and compounds (ligands) were used as sources of computer-based modeling. A multitude of databases of compounds were used as sources of chemical structures in one or more of the highlighted studies including ZINC (Rout et al., 2015; Rout and Mahapatra, 2016; Verma et al., 2015), ChEMBL-NTD (Sullivan et al., 2015), DrugBank (Ren et al., 2016; Rout et al., 2015; Rout and Mahapatra, 2016), PubChem (Kumari et al., 2016; Rout and Mahapatra, 2016), and ChemSpider (Rout and Mahapatra, 2016). For antimalarial protein targets, the databases applied included PlasmoDB 9.2 (Lhouvum et al., 2013; Pandey et al., 2014; Rout et al., 2015; Sharma et al., 2016), PFAM (Pandey et al., 2014; Rout et al., 2015), CDD [29], ZINC (Lhouvum et al., 2013; Pandey et al., 2014), and InterPro (Campbell et al., 2014; Rout et al., 2015). Protein structures were downloaded from these databases and validated using RAMPAGE (Paul et al., 2015; Sharma et al., 2016), PROCHECK (Bouillon et al., 2013; Campbell et al., 2014; Kruggel and Lemcke, 2009; Lhouvum et al., 2013; Mehra et al., 2015; Paul et al., 2015; Raza et al., 2017; Rout et al., 2015; Rout and Mahapatra, 2016), Verify-3D (Campbell et al., 2014; Lhouvum et al., 2013; Rout et al., 2015; Rout and Mahapatra, 2016; Sharma et al., 2016), ERRAT (Campbell et al., 2014; Lhouvum et al., 2013; Rout et al., 2015; Rout and Mahapatra, 2016; Sharma et al., 2016), CHIMERA (Paul et al., 2015), and PROSA (Rout et al., 2015; Rout and Mahapatra, 2016; Sharma et al., 2016; Verma et al., 2015). Stability check was performed using Gromacs (Lhouvum et al., 2013; Mehra et al., 2015; Paul et al., 2015; Rout and Mahapatra, 2016; Sharma et al., 2016).
Lead identification/optimization
Most studies applied in silico modeling for structure-based or ligand-based design for potential antimalarial candidates using known targets specific to Plasmodium. Inosine monophosphate dehydrogenase (IMPDH) is an important enzyme in the purine biosynthesis of P. falciparum. A 3D homology model for this parasite enzyme was made using human IMPDH (PDB code 1NF7) as a template (Raza et al., 2017). The in silico combinatorial library of 1,347 ribavirin derivatives was designed and virtually screened. Finally, 5 ligands were shown to be more specific to P. falciparum IMPDH than human IMPDH II. Proteasome inhibitors have been demonstrated as potential antimalarial compounds with action on asexual and sexual Plasmodium blood stages. Various proteasome inhibitors with potent antimalarial activity and low host cytotoxicity were designed and evaluated based on the carmaphycin B scaffold (LaMonte et al., 2017). Cell-based and Plasmodium proteasome assays revealed one promising compound (compound 18) active against both asexual blood stages and gametocytes. In vitro evolution in S. cerevisiae, biochemical assays, and molecular modeling studies confirmed that this activity is due to specific inhibition of the β5 subunit of the proteasome. Plasmodium lipid kinase PI(4)K (phosphatidylinositol-4-OH kinase) is a ubiquitous eukaryotic enzyme that phosphorylates lipids to regulate intracellular signaling and trafficking. Virtual screening for inhibitors of this enzyme as potential antimalarial drugs was performed (Ren et al., 2017).
A homology modeling of PI(4)K from P. falciparum was initially built. The compound KAI715 which showed high potent activity and selectivity for PI(4)K was docked into the enzyme ATP-binding site. The optimized pharmacophore modeling of this target-ligand complex (HyopA) was then used to search a large chemical database. All drug-like hit compounds with satisfactory ADMET properties that passed the pharmacophore-based virtual screening were identified using Lipinski’s rule of five (Lipinski et al., 2016) and ADMET (aqueous solubility, human intestinal absorption, penetration across blood-brain barrier (BBB), cytochrome P450 2D6 inhibition, hepatotoxicity, and plasma-protein binding) prediction. The molecular docking method was then further carried out to re-filter these screened compounds.Two more potent compounds in addition to KAI715 were designed. DegP protein has been shown to be involved in regulation of thermo-oxidative stress generated during asexual life cycle of Plasmodium. The protein is probably required for survival of parasite in host. A 3D structure of PfDegP was generated using MODELLER based on PlasmoDB and protein database bank (PDB). The model was validated using RAMPAGE and ERRAT. An in silico screening of small molecule database against PfDegP was performed using Glide and molecular dynamics simulation of protein and protein-ligand complex was carried out using GROMACS. Seven lead compounds which were inhibitors of PfDegP were finally generated (Sharma et al., 2016).
P. falciparum M18 aspartyl aminopeptidase (PfM18AAP) is only aspartyl aminopeptidase which is found in the Plasmodium genome and is essential for its survival. This enzyme performs various functions in the parasite and the erythrocytic host such as hemoglobin digestion, erythrocyte invasion, parasite growth, and parasite escape from the host cell. The 3D-QSAR modeling, pharmacophore modeling, and molecular docking were employed to identify novel potent inhibitors of PfM18AAP (Kamuri et al., 2016). Ten novel PfM18AAP inhibitors from ChEMBL antimalarial library, 2 novel inhibitors from each derivative of quinine, chloroquine, 8-aminoquinoline, and 10 novel inhibitors from WHO antimalarial drugs were used. P. vivax leucine amino peptidase (M17 PvLAP) belonging to the metallo-aminoeptidase family, plays a significant role in the catalysis of the terminal stage of hemoglobin degradation which is essential for growth and development of P. vivax (Lee et al., 2010). A homology model was generated using MODELLER and applied various in silico methods such as structure based, ligand based and de novo drug designing to design potential compounds that are inhibitors of these enzymes (Rout and Mahapatra, 2016). Out of the ten potential candidates, 2-[(3-azaniumyl-2-hydroxy-4-phenylbutanoyl) amino]-4-methylpentanoate was identified as the best inhibitor in terms of docking score and pharmacophoric features. The reliability of the binding mode of the inhibitor was confirmed by molecular dynamics (MD) simulation study with GROMACS software. Finally, in silico ADMET properties were evaluated.
P. falciparum glycogen synthase kinase-3 (PfGSK-3) is one of the eukaryotic protein kinases that have been identified as essential for Plasmodium. Although the physiological functions of PfGSK-3 are still unknown, it has been suggested as a putative target for novel antimalarial drugs. Homology modeling and molecular docking and 10 different HsGSK-3b templates were applied for the model building of PfGSK-3 (Kruggel and Lemcke, 2009). The evaluated top models were used to compile an ensemble of PfGSK-3 models and for the structure-based design of potential ATP-binding site inhibitors of PfGSK-3. Thieno[2,3-b]pyridines were identified as a new class of PfGSK-3 inhibitors. P. falciparum CTP synthetase (CTPS), choline kinase (CK), and glutathione S-transferase (GST) selected based on their connectedness and functional importance in biochemical/metabolic pathways of P. falciparum, were used in virtual screening of ZINC database entries which led to the design and synthesis of arylsulfonyloxy acetimidamides as their inhibitors (Verma et al., 2015). Peptidyl-prolyl cis/trans isomerases (PPIases) constitute an enzyme superfamily that converts cis and trans amide bonds in proteins and peptides, and in addition, cellular processes such as apoptosis or protein synthesis in almost all living cells. Fk506-binding proteins (FKBPs) are the largest and most varied of the PPIases. Through application of molecular docking, it was demonstrated that the substrates ILS-920 and WYE-592 bind less-favorably with human FKBP12 (hFKBP12) and P. falciparum FKBP35 (PfFKBP35) compared to a competing substrate rapamycin (MacDonald and Boyd, 2015).
Quantum-similarity approach was applied for discovery of novel liver-stage antimalarials (Sullivan et al., 2015). Testing of only five of the model-predicted compounds in vitro and in vivo hepatic stage drug inhibition assays with P. berghei identified four novel chemical structures. All inhibited liver stage Plasmodium at a single oral dose in the quantitative PCR mouse liver-stage sporozoites-challenge model. Cethromycin (ABT-773), a macrolide-quinoline hybrid, was also identified as a potential candidate for further development due to its extensive safety profile. P. falciparum phosphatases are important elements of intraerythrocytic development expressed throughout the life cycle. P. falciparum Mitogen-Activated Protein Kinase (MAPK) phosphatase (MKP) subgroup signaling cascades are critical components of sexual stage proliferation. Interaction of the MKP with its phosphoprotein substrate depends on three conserved residues in the consensus atypical dual-specificity phosphatase (DUSP) domain binding pocket. A homology model of the atypical dual-specificity phosphatase (DUSP) domain was developed for use in high-throughput in silico screening of the available library of antimalarial compounds from ChEMBL-NTD (Campbell et al., 2014). Seven compounds were selected for further evaluation of antimalarial activity in vitro. Out of these, 4 compounds (390097, 524725, 525841, and 585222) showed promising activity against C9 parasites. The cytochrome bc1 complex (ubiquinol: cytochrome c oxidoreductase, respiratory Complex III) is a key enzyme of the mitochondrial electron-transfer chain in all metazoa and protozoa including Plasmodium. It catalyzes the transfer of electrons from ubiquinol to cytochrome c. The inhibition of cytochrome bc1 blocks the mitochondrial respiratory chain and the consequent arrest of pyrimidine biosynthesis, which is essential for parasite development.
The 2D- or 3D-SAR was developed and a docking analysis was conducted for a series of 4(1H)-quinolones as cytochrome bc1 inhibitors (Villalobos et al., 2013). The substituents R1 and R4 in 4(1H)-quinolones analogues were key modulators to enhance the antimalarial activity. The appropriate binding conformations and orientations of these compounds interacting with cytochrome bc1 were also revealed by molecular docking. Eight promising compounds were designed and presented as reference compounds for synthesis and antimalarial evaluation. Subtilisin-like serine protease SUB1 of Plasmodium merozoites plays a dual role in egress from and invasion into host erythrocytes. The P. vivax-SUB1 (PvSUB1) was characterized and shown to display similar cellular location, auto-processing, and enzymatic properties as its PfSUB1 ortholog (Bouillon et al., 2013). Homology modeling and molecular docking were applied to search for potential PvSUB1 inhibitors using 3D models of PvSUB1 as targets. The 306 best predicted hits were selected and tested for their inhibitory potency on the PvSUB1 recombinant enzyme. Active compounds were then tested for their antimalarial activity in vitro. Five most promising compounds particularly compound 2 were shown to exhibit specific activity against P. falciparum merozoite egress and invasion in P. berghei-infected mice.
Target identification/validation
A number of Plasmodium targets were identified and validated by application of in silico modeling approach. interstitial cell adhesion molecule- (1ICAM-1) belongs to the immunoglobulin-like superfamily and plays an important role in cell recognition, cell adhesion and cell aggregation. ICAM-1 has been identified as an endothelial receptor for P. falciparum infected red blood cells (IRBCs). Cytoadherence of parasitized red cells is mediated by PfEMP1 (P. falciparum erythrocyte membrane protein 1) expressed on the surface of IRBCs. Cytoadherence of ICAM-1 to surface expressed antigen on IRBCs leads to cascade of consequences that contribute to the development of cerebral malaria. IRBC-endothelial cells cytoadherence therefore represents a promising target to attenuate or eliminate down-stream pathophysiological events. Target identification/validation was applied to understand the forces operating between ICAM-1 and PfEMP1 cytoadherence complex and designing better cytoadherence peptides which could be useful for development of potential anti-adhesion therapeutics (Mehra et al., 2015). P. falciparum NMT (PfNMT) is responsible for the sexual blood stages of the parasite and is essential for transmission. The 3D-structure of PfNMT was modeled using Modeler (v.9.0) taking P. vivax NMT (PvNMT) as the template (Paul et al., 2015).
The generated structure was then validated using various programs such as PROCHECK, RAMPAGE server, and CHIMERA and the stability of the model was checked by Gromacs 5.0. The enzyme was shown to be a vital target and the modeled structure could be further applied in molecular docking studies for novel drug design. A search for unique proteins was conducted by a comparative genomics study (Rout et al., 2015). Eleven proteins (phosphoenolpyruvate carboxykinase, pyridoxine/pyridoxal 5-phosphate biosynthesis enzyme, beta-hydroxyacyl-ACP dehydratase, NADH dehydrogenase, fumarate hydratase, phosphoenolpyruvate carboxylase, putative uncharacterized protein, uncharacterized protein involved in lipopolysaccharide biosynthesis, RNA pseudouridylate synthase, pseudouridine synthase, and lipoate–protein ligase) were prioritized as antimalarial drug targets. The homology models of two uncharacterized proteins were built using MODELLER (v9.13) software from possible templates. Functional annotation of these proteins was done by the InterPro databases and ProBiS server by comparison of predicted binding site residues. The model was subjected to in silico docking study to search for potent lead compounds from the ZINC database by Dock Blaster software using AutoDock 4.
Results from this study would facilitate the selection of proteins and putative inhibitors for entry into drug design production pipelines. Six Plasmodium specific phosphatases and 33 putative phosphatases with absence of human orthologs were identified (Pandey et al., 2014). Using phylogenetic analysis of the blood-stage asexual cycle, the enzymes were identified as atypical MAPK phosphatases. This has led to the identification of the putative phosphatase from PF3D7_1305500 as an important element of intraerythrocytic development expressed throughout the life cycle (35). Additional bioinformatics analysis delineated a conserved signature motif and three residues with potential importance to functional activity of the atypical dual-specificity phosphatase (DUSP) domain. PFI1625c was identified as a putative metalloprotease present in Plasmodium genome (Lhouvum et al., 2013). Probing PFI1625c active site with 199 different peptides from a combinatorial peptide library indicated preference of PFI1626c toward hydrophobic residue substituted peptides. The peptide P550 (LVIVAKRA) was shown to exhibit significantly better interaction within the active site than a template peptide (LSRVAKRA). The molecular dynamic’s simulation studies confirmed integrity of the complex. The structural and biochemical differences between PFI1625c with human metalloprotease suggest that the enzyme could be exploited as drug targets for future antimalarial development.
In silico modeling is a useful tool that plays important role in identification/validation of potential antimalarial drug targets as well as identification of potential lead candidates during drug discovery phase. In addition, it is also useful in elucidating their mechanisms of action and their potential clinical efficacy. Exploring potential drugs via computational modeling is a safe, frugal, and effective method to discover, develop, or repurpose potential treatments. Nevertheless, the antimalarial activity of these potential candidate compounds need to be further confirmed by nonclinical and clinical validation using a series of in vitro and in vivo assays. Multi-targeting approaches have the best potential to be the most effective. In addition, the in silico approach should also be applied in parallel for the prediction of drug-like and ADME properties of the candidate molecules.
The authors have not declared any conflict of interests.
REFERENCES
|
Bouillon A, Giganti D, Benedet C, Gorgette O, Pêtres S, Crublet E, Girard-Blanc C, Witkowski B, Ménard D, Nilges M, Mercereau-Puijalon O (2013). In silico screening on the three-dimensional model of the Plasmodium vivax SUB1 protease leads to the validation of a novel anti-parasite compound. J. Biol. Chem. 288(25):18561-18573.
Crossref
|
|
|
|
Campbell CO, Santiago DN, Guida WC, Manetsch R, Adams JH (2014). In silico characterization of an atypical MAPK phosphatase of Plasmodium falciparum as a suitable target for drug discovery. Chem. Biol. Drug Des. 84(2):158-168.
Crossref
|
|
|
|
|
Hodos RA, Kidd BA, Shameer K, Readhead BP, Dudley JT (2016). In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 8(3):186-210.
Crossref
|
|
|
|
|
Kumari M, Chandra S, Tiwari N, Subbarao N (2016). 3D QSAR, pharmacophore and molecular docking studies of known inhibitors and designing of novel inhibitors for M18 aspartyl aminopeptidase of Plasmodium falciparum. BMC Struct. Biol. 16(1):12-20.
Crossref
|
|
|
|
|
LaMonte GM, Almaliti J, Bibo-Verdugo B, Keller L, Zou BY, Yang J. (2017). Development of a potent inhibitor of the Plasmodium proteasome with reduced mammalian toxicity. J. Med. Chem. 60(15):6721-6732.
Crossref
|
|
|
|
|
Lhouvum K, Ramakrishnan V, Trivedi V (2013). Insight into structural and biochemical determinants of substrate specificity of PFI1625c: correlation analysis of protein-peptide molecular models. J. Mol. Graph. Model. 43:21-30.
Crossref
|
|
|
|
|
Lipinski CA (2016). Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 101:34-41.
Crossref
|
|
|
|
|
Lounnas V, Ritschel T, Kelder J, McGuire R, Bywater RP, Lounnas V, Ritschel T, Kelder J, McGuire R, Bywater RP, Foloppe N (2013). Current progress in structure-ased rational drug design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 5:e201302011.
Crossref
|
|
|
|
|
MacDonald CA, Boyd RJ (2015). Molecular docking study of macrocycles as Fk506-binding protein inhibitors. J. Mol. Graph. Model. 59(Suppl. C):117-122.
Crossref
|
|
|
|
|
Maier JKX, Labute P (2014) Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins: Structure, Function, and Bioinformatics 82(8):1599-1610.
Crossref
|
|
|
|
|
Margolis R, Derr L, Dunn M, Huerta M, Larkin J, Sheehan J, Guyer M, Green ED (2014). The National Institutes of Health's Big Data to Knowledge (BD2K) initiative: capitalizing on biomedical big data. J. Am. Med. Inform. Assoc. 21(6):957-958.
Crossref
|
|
|
|
|
March-Vila E, Pinzi L, Sturm N, Tinivella A, Engkvist O, Chen H, Rastelli G (2017). On the integration of in silico drug design methods for drug repurposing. Front. Pharmacol. 8:298-306.
Crossref
|
|
|
|
|
Mehra A, Jerath G, Ramakrishnan V, Trivedi V (2015). Characterization of ICAM-1 biophore to design cytoadherence blocking peptides. J. Mol. Graph. Model. 57(Suppl. C):27-35.
Crossref
|
|
|
|
|
Méndez-Lucio O, Naveja JJ, Vite-Caritino H, Prieto-Martínez FD, Medina-Franco JL (2016). Review. One Drug for Multiple Targets: A Computational Perspective. J. Mex. Chem. Soc. 60(3):168-181.
|
|
|
|
|
Paul P, Chowdhury A, Das Talukdar A, Choudhury MD (2015). Homology modeling and molecular dynamics simulation of N-myristoyltransferase from Plasmodium falciparum: An insight into novel antimalarial drug design. J. Mol. Model. 21(3):37-43.
Crossref
|
|
|
|
|
Pandey R, Mohmmed A, Pierrot C, Khalife J, Malhotra P, Gupta D (2014). Genome wide in silico analysis of Plasmodium falciparum phosphatome. BMC Genom. 15(1):1024-1030.
Crossref
|
|
|
|
|
Peltenburg H, Groothuis FA, Droge ST, Bosman IJ, Hermens JL (2013). Elucidating the sorption mechanism of "mixed-mode" SPME using the basic drug amphetamine as a model compound. Anal. Chem. Acta 782:21-27.
Crossref
|
|
|
|
|
Raza M, Khan Z, Ahmad A, Raza S, Khan A, Mohammadzai IU. (2017). In silico 3-D structure prediction and molecular docking studies of inosine monophosphate dehydrogenase from Plasmodium falciparum. Comput. Biol. Chem. 71:10-19.
Crossref
|
|
|
|
|
Ren JX, Gao NN, Cao XS, Hu QA, Xie Y (2016). Homology modeling and virtual screening for inhibitors of lipid kinase PI(4)K from Plasmodium. Biomed. Pharmacother. 83:798-808.
Crossref
|
|
|
|
|
Rout S, Warhurst DC, Suar M, Mahapatra RK (2015). In silico comparative genomics analysis of Plasmodium falciparum for the identification of putative essential genes and therapeutic candidates. J. Microb. Methods 109 (Suppl. C):1-8.
Crossref
|
|
|
|
|
Rout S, Mahapatra RK (2016). In silico screening of novel inhibitors of M17 leucine amino peptidase (LAP) of Plasmodium vivax as therapeutic candidate. Biomed. Pharmacother. 82:192-201.
Crossref
|
|
|
|
|
Sharma D, Soni R, Patel S, Joshi D, Bhatt TK (2016). In-silico studies on DegP protein of Plasmodium falciparum in search of anti-malarials. J. Mol. Model. 22(9):201-211.
Crossref
|
|
|
|
|
Sullivan DJ, Liu Y, Mott BT, Kaludov N, Martinov MN (2015). Discovery of Novel Liver-Stage Antimalarials through Quantum Similarity. PLoS ONE 10(5):e0125593.
Crossref
|
|
|
|
|
Verma S, Debnath U, Agarwal P, Srivastava K, Prabhakar YS (2015). In silico exploration for new antimalarials: arylsulfonyloxy acetimidamides as prospective agents. J. Chem. Info. Model. 55(8):1708-1719.
Crossref
|
|
|
|
|
Villalobos TP, Ibarra R, Acosta JJ (2013). 2D, 3D-QSAR and molecular docking of 4(1H)-quinolones analogues with antimalarial activities. J. Mol. Graph. Model. 46:105-124.
Crossref
|
|
|
|
|
Webb B, Sali A (2014). Protein structure modeling with MODELLER. Methods Mol. Biol. 1137:1-15.
Crossref
|
|
|
|
|
World Health Organization (WHO) (2015). World Malaria Report 2015. World Health Organization: Geneva, Switzaerland.
|
|