ABSTRACT
Genetic diseases are poorly reported in sub-Saharan Africa, especially in Burkina Faso. The reasons for this reality are multifactorial including, the difficulty of diagnostic confirmation, financial accessibility and even the difficulty with referral of cases by medical staff. Genetic diseases, although relatively rare, exist in families and deserve special attention in our paraclinical assessments. Few cases can be diagnosed in sub-Saharan Africa. The clinical case we are reporting here is a dystrophynopathy by mutation of the Duchenne muscular dystrophy (DMD) gene. This is the first confirmed detection of a frameshift mutation in the DMD gene in a boy received at Saint Camille Hospital in Ouagadougou, Burkina Faso. This is a 13-year-old MR boy, from a family of four siblings, all male, from the eastern region of Burkina Faso. The boy had a clinical picture of myopathy with difficulty in walking, frequent falls, myogenic syndrome with stool sign, Gowers sign and scapula alata, all leading to a suspicion of dystrophinopathy with a request for genetic analysis. The DMD gene responsible for the disease is located on the X chromosome (Xp21.2-p21.1). The study of the dystrophin gene (DMD) was done using three methods, namely MLPA, high throughput sequencing and Sanger sequencing. The results led to the identification of a frameshift mutation of exon 71 in the DMD gene: it is a hemizygotic variant with ribosomal shift of the DMD gene NM_004006.2 (DMD): c.10258del p.(Ser3420Leufs*25). This clinical case led for the first time in Burkina Faso to the confirmed diagnosis of hereditary muscular dystrophy resulting from a mutation with a frameshift in exon 71 of the DMD gene in the hemizygotic state in a 13-year-old boy, a student and the eldest sibling of 4 boys, three of whom have myopathy.
Key words: Mutation, Duchenne muscular dystrophy (DMD) gene, dystrophinopathy, duchenne versus becker, case report.
Abbreviation:
MR, Malo RABO (pseudonym of the patient); DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; MLPA, Multiplex-ligation dependent probe amplification; CPAD, Complex of dystrophin-associated proteins; CK, Creatine kinase; AST, Aspartate aminotransferase; ALT, Alanine aminotransferase; IVC, interventricular communication; CERBA, Pietro Annigoni Biomolecular Research Center; LABIOGENE, Laboratory of Molecular Biology and Genetics; HOSCO, Saint Camille Hospital of Ouagadougou; USTA, University Saint Thomas d’Aquin.
The frame shift mutation of the DMD gene is rarely reported in the literature in patients with dystrophinopathy. This is the first detection of a mutation with ribosomal shift in the DMD gene in a patient from Burkina Faso. This clinical case aims to report the existence of a rare mutation of the DMD gene in sub-Saharan Africa.
Duchenne and Becker muscular dystrophies (DMD and BMD, respectively) result from mutations in the DMD gene (MIM #300377) encoding a membrane cytoskeleton protein, dystrophin. These are genetic diseases linked to sex chromosomes X and variable clinical expression in women (usually healthy carriers) due to random inactivation of the X chromosome while male hemizygotes manifest the disease. It is transmitted recessively in women and dominant in men.
The alteration on the dystrophin sequence resulting in a total absence or partial deficiency (truncated protein and/or in decreased quantity) in dystrophin has consequences that are expressed in several ways. In skeletal muscles, there is a loss of membrane integrity of the muscle fibers responsible for muscular dystrophy (Muntoni et al., 2003; Straathof et al., 2015) while in cardiac muscle abnormal expression of dystrophin is responsible for isolated cardiomyopathy or most often associated with muscular dystrophy (Deburgrave et al., 2007; Ortez et al., 2019). On the other hand, cardiomyopathies linked to the absence of dystrophin can be noted both in DMD and/or BMD patients and in women who are said to transmit these pathologies (Bushby et al., 1993; Mori-Yoshimura et al., 2018).
DMD is clinically characterized by progressive muscle weakness, with an incidence of 1 in 3500 to 6000 men (Gospe et al., 1989). Similarly, BMD is characterized by high phenotypic variability, ranging from severe muscle weakness with cardiomyopathy and early death to exercise-induced myalgia and/or muscle cramps associated with an increase in serum creatine kinase (CK) activity (Bakker et al., 1997; Melacini et al., 1996; Rahimov and Kunkel, 2013; Kwiatkowska et al., 2020). However, cardiomyopathy has been found in BMD patients without skeletal muscle weakness (Serrano and Munoz-Canoves, 2017). These two genetic diseases (BMD and DMD) are caused by the mutation of the DMD gene, which codes for a protein called dystrophin, which is essential for structural muscle stability. Genetic mutations in the DMD gene are very heterogeneous and can be deletions (65%), duplications (5.10%) and point mutations (10 - 15%). The DMD gene is subject to high proportion of neo mutations. The DMD gene, located on the short arm of the X chromosome (Xp21.2-p21.1), is the largest human gene known to date. With a size of 2.2 million base pairs, it occupies 0.1% of the human genome and 1.5% of the X chromosome sequence. The 14 kilobase (kb) messenger RNA is composed of 79 exons that constitute only 0.6% of the overall gene sequence (Nakamura et al., 2015).
Major genomic rearrangements such as deletions or duplications of one or more exons are in the majority (70% of cases), and point mutations are found in 20 to 30% of cases (Gospe et al., 1989; Cripe and Tobias, 2013; Buddhe et al., 2018). The severity of the phenotype (DMD versus BMD) can be correlated with the impact of the mutation on dystrophin expression in more than 90% of cases. Exceptions to this so-called reading framework rule result from various mechanisms and are observed in less than 10% of cases. In addition to the benefit of having a confirmed diagnosis to the patient, identification of the mutation allows genetic counselling adapted to the family, and a potential inclusion of the patient in therapeutic trials based on the genotype (Gospe et al., 1989; Straathof et al., 2015).
At present, there is no curative treatment. Corticosteroids, currently indicated in the early stages of the disease, are the only ones that have shown a change in the natural history of the disease, independent of the genetic mutation cases (Cripe and Tobias, 2013).
Demographic details
Our patient is a 13-year-old boy from a family of four children, all male, from the eastern region of Burkina Faso. The patient is a student in the 5th grade.
Medical history
The patient had a history of normal birth and psychomotor development until the age of 11 years when the first symptoms appeared and motivated a medical consultation. The first signs were of difficulty walking, and the physical examination had not noted any visible deformation. The hypothesis of sickle cell disease was raised and hemoglobin electrophoresis was requested resulting in the detection of AC genotype.
He had a second medical consultation two months after the first one with the reason being difficulty walking and fatigability for 1 month. The physical examination noted:
Good general condition, normal colored and anicteric mucous membranes, decreased motor strength in the lower limbs with motor strength (FM) = 3/5 in both limbs and predominant in the thigh muscles. No sensory disturbances. Osteotendinous reflexes (ROT) were reduced. The diagnostic differential of myopathy and neuropathy was made. The patient was referred to neurology department.
About ten days later, a medical consultation was conducted for progressive weakness of the lower limbs associated with frequent falls. The examination notes a good general condition, a myogenic syndrome with stool sign, Gouers sign, scapula alata. The diagnosis of limb-girdle muscular dystrophy was hypothesized. Paraclinical examinations were requested: CK = 6351 U/L with standards between 80 and 200 U/L (i.e. more than 30 times normal), ALT = 167 U/L, AST = 102 U/L, creatinine = 40.84 micromol/L,hemogram shows a white blood cell count at 6090 cells/µL; hemoglobin level = 12.4 g/dL; platelet count = 441000/µL. The electrocardiogram, echocardiogram, electroneuromyogram, and blood ionogram were without particular abnormality. The medical prescription was Défal cp 30 mg ½ cp per day in the morning; Kaleoride cp 600 mg ½ cp at noon; Albendazole cp 1cp/jr for 3 days; Prazol capsules 20 mg 1 capsule in the morning and a low sodium diet. There was no secondary effect related to drug treatment.
Under this treatment, the child during neurological follow-up and CK monitoring had elevated CK levels at 8171 U/L; AST at 109.3 U/L; ALT at 130 U/L. This high CK level motivated the consultation, which found a myogenic syndrome with stool sign, Gowers sign and scapula alata with the notion of difficulty walking, frequent falling and fatigability. The cardiac Doppler echo was within normal limits. We therefore continued with a study of the DMD gene in search of a mutation. The request for the study of the DMD gene was preceded by a family interview looking for events related to dystrophinopathy in the maternal family. The child was put under kinesitherapy sessions.
Family history
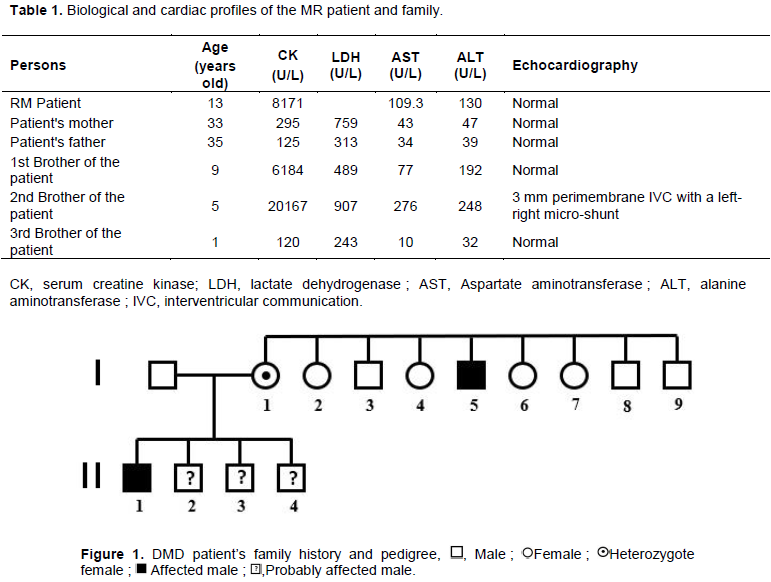
There is a notion of similar undocumented disorders in our patient's maternal uncle (one of the brothers of the MR patient's mother's mother). This uncle was said to have had difficulty walking, frequent falls with an inability to recover resulting in disability at the age of 30 years. Indeed, the mother of the MR patient comes from a sibling of 9 children, including 4 boys (including the uncle who has difficulty getting up) and 5 girls (including the patient's mother). The MR patient's mother's biological test scores found CK at 295 U/L and LDH at 759 U/L. While the CK of the MR patient's father had 125 U/L and LDH = 313 U/L. Of the three brothers of the MR patient, respectively aged 9, 5 and 1 year old, two had high CK values (Table 1). The 5-year-old boy with high CK (20167 U/L) also had a small 3 mm perimembrane interventricular communication (IVC) on the cardiac Doppler with a left-right micro-shunt without impact on the cardiac chambers (Table 1). The latter with a very high CK value was placed on the same treatment as the MR patient.
The pedigree of the parents and the MR patient is shown in Figure 1.
Genetic analysis of the DMD gene in the MR patient
Genomic DNA was extracted from peripheral blood leucocytes according to standard protocols.
The identification of the mutation of the hemizygous frame shift variant of the DMD gene NM_004006.2(DMD): c.10258del p. (Ser3420Leufs*25) in our MR patient was performed by three molecular biology and genetics techniques such as Multiplex Ligation-dependant Probe Amplification (MLPA), High throughput sequencing using next-generation sequencing (NGS) and confirmation of the DMD variant by targeted Sanger sequencing; at the Biochemistry and Molecular Biology Laboratory Grand Est of the University Hospital of Lyon in France. The first step consisted of the search for the most frequent lesions: deletions and duplications of exons using MLPA.
Multiplex ligation-dependant probe amplification:
This technique allows the number of copies for each of the 79 exons to be estimated using standardized probes that highlight both deletions and duplications. This fast and reliable technique using SALSA KIT P034/P035 MRC-Holland, is one of the most commonly used in routine, both for index cases and for the status search of women at risk: it solves the majority of quantitative anomalies on the DMD gene. DNA quality can sometimes be a limiting criterion for good interpretation and the anomaly of an isolated exon must always be controlled by another technique (Muntoni et al., 2003).
The MLPA analysis initially performed in our MR patient did not detect any duplication or deletion of the DMD gene. Then high throughput sequencing was performed in search of mutations.
High throughput sequencing
This technique was performed using preparation of the Kappa Nimblegen library and Illumina NextSeq™500 Sequencing System (at a reading length of 2 × 150 bp). Considering the family medical history of MR, a panel of genes was compiled, which have been reported in literature to be linked to muscular dystrophy: Limb-girdle muscular dystrophy (LGMD) is a genetically and clinically heterogeneous group of rare muscular dystrophies. The list of genes in the "LGMD main genes" panel considered was: ANOS, CAPN3, CAV3, DMD, DYSF, FKRF, SGCA, SGCG.
Computer analysis Home-made Pipeline "Papillyon": produced sequences aligned and compared to the human reference genome (GRCh37/hg19). Targeted analysis of the DMD gene (NM_004006.2) was done. The intronic exons and borders of the 79 exons of the DMD gene are covered with a depth greater than 30 X. This high throughput sequencing analysis allowed us to detect in our MR patient, a hemizygotic frameshift variant of the DMD gene NM_004006.2 (DMD): c.10258del p. (Ser3420Leufs*25) located on exon 71. This variant is absent from LOVD but has been reported in the literature in a patient with severe Becker phenotype (Straathof et al., 2015; Mornet and Rivier, 2017).
The confirmation of the DMD variant was done by targeted Sanger sequencing. The pathogenic variant was confirmed by conventional dideoxy sequencing using Big-Dye Terminators (Life Technologies) after specific amplification. Sanger sequencing is a long and costly but effective technique, serving as a reference technique to validate results provided by new high throughput sequencing techniques. Sanger sequencing: Amplification by the "classical" PCR technique then bidirectional capillary sequencing on ABI 3500xL. The sequences produced were compared with the human reference genome (GRCh37/hg19) with SeqScape v3, Life Technologies software. The search for mutation of the DMD gene in the MR patient was positive with the identification of a hemizygous frameshift variant of the DMD gene NM_004006.2(DMD): c.10258del p. (Ser3420Leufs*25) located on exon 71. The discovery of this variant made it possible to diagnose dystrophynopathy in the MR patient (Table 2). A muscle biopsy was not obtained to determine the exact phenotype (DMD or BMD) by immunohistochemistry and Western blot. No Adverse and unanticipated events were noted.

Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are monogenic X-linked recessive disorder and are both located on Xp21.2-p21.1, the short (p) arm of the X chromosome between positions 21.2 and 21.1 (OMIM*300377).
Cases of genetic diseases are underestimated in sub-Saharan Africa due to a lack of access to diagnostic means of confirmation. The case of our patient, who is a young boy, a student (with normal cognitive development), clinically presenting a myopathy with a suspicion of Duchenne or Becker dystrophynopathy, could have gone unnoticed for the confirmation of the effective existence of a mutation of the DMD gene. In addition, a rare mutation has been identified in our patient. The MLPA analysis had not noted any deletion or duplication; and it was the high throughput sequencing that made it possible to detect this rare mutation, a frameshift variant of the DMD gene in the hemizygous state located on exon 71, considered pathogenic and part of the new mutations of the DMD gene (Mornet and Rivier, 2017).
This variant is absent in the Leiden Open Variation database (LOVD) but has been reported in the literature in a patient with a Severe Becker phenotype and normal cognitive development, but a muscle biopsy could not be performed for the RNA study (Mornet and Rivier, 2017). In addition, Straathof et al. found this pathogenic novel mutation: a frameshift mutation in exon 71 (c.10258del, p.ser3420-Leufs25*) in one of their patients who also had a severe BMD phenotype with normal cognitive development (Straathof et al., 2015).
This genotype/phenotype discrepancy could be explained by a catch-up of the exon 71 skipping reading frame on some of the transcripts. This mechanism has been reported for some frameshift variants in exon 71. These are frameshift mutations c.193delG and c.4922insT in two patients with BMD in whom muscle biopsy followed by Western blot shows a slightly reduced size of the dystrophin protein cases (Cripe and Tobias, 2013).
The deletion is frame shift, so we expect to have a DMD profile with the absence of the dysthrophin protein. But with regard to the clinic, particularly the age at which symptoms begin, such as difficulty walking (after the age of 10 years), the phenotype would be that of a BMD with partial production of dystrophin. The consequences and life expectancy are not the same for these two forms, hence the indication of a muscle biopsy is necessary to affirm DMD or BMD. In our MR patient we did not obtain a muscle biopsy to determine the exact phenotype (DMD or BMD) by immunohistochemistry and Western blot. The MR child was put on kinesitherapy and cardiac, respiratory and Achilles tendon monitoring is required. The mother of the MR patient is probably a carrier and the other children will have the same follow-up.
Dystrophin is one of the largest proteins in the body, located on the inner surface of the skeletal muscle fibre, where it provides a structural link between the cytoskeleton and the sarcolemma, via a set of proteins called the "dystrophin associated protein complex" (CPAD) (Péréon et al. 2015).
The clinical signs of myopathy noted in our patient are explained by the involvement of the dystrophy gene. The mechanical contraction action of muscle fibers generates micro lesions of the sarcolemma, in the absence of the link between the muscle fiber membrane and myofibrils represented by dystrophin and CPAD (Campbell, 1995; Petrof et al., 1993). However, we did not observe any cardiac muscle abnormalities or cognitive impairment in the MR patient.
It is established that in Duchenne muscular dystrophy (DMD), not only is there a lack of dystrophin, but the expression of the proteins that make up the CPAD is also much reduced. The excessive fragility of the membrane with respect to mechanical stress and the increase in its permeability, the dysregulation of calcium homeostasis, the disturbances of NO synthase, oxidative stress, all of which result from the loss of dystrophin, are at the origin of muscle necrosis, followed by an initially effective regeneration. Over time, it becomes exhausted, inflammation and endomysial fibrosis set in, with the progressive replacement of muscle fibres by fibroadipular tissue and the loss of muscle function (Péréon et al., 2015).
Genetic counselling was offered and carried out to the family of the MR patient with multidisciplinary follow-up. This clinical case reports dystrophinopathy (Duchenne DMD versus Becker BMD) for which the confirmatory genetic diagnosis is not available in Burkina Faso. This is the first detection of a rare mutation of the DMD gene to Frameshift type mutation in a 13-year-old boy (from a sibling of four boys, three of whom have biological signs of myopathy) from Burkina Faso, at Saint Camille Hospital in Ouagadougou.
This potentially fatal mutation was detected following high throughput sequencing and confirmed by Sanger sequencing. This genetic disease requires a multidisciplinary team and the appropriate treatment is still being investigated.
The authors would like to thank Dr Rita Menassa and Dr Laurence Michel-Calemard of the Centre Hospitalier Universitaire de Lyon, Department of Biochemistry and Molecular Biology Grand Est, GHE - CBPE - 59 Boulevards Pinel 69677 Bron Cedex-France for the sequencing performed. They also thank Dr Dorcas Obiri-Yeboah and the staff of the Saint Camille de Ouagadougou Hospital (HOSCO) for their contribution to the medical care of this patient.
The authors have not declared any conflict of interests.
MR, Malo RABO (pseudonym of the patient); DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; MLPA, Multiplex-ligation dependent probe amplification; CPAD, Complex of dystrophin-associated proteins; CK, Creatine kinase; AST, Aspartate aminotransferase; ALT, Alanine aminotransferase; IVC, interventricular communication; CERBA, Pietro Annigoni Biomolecular Research Center; LABIOGENE, Laboratory of Molecular Biology and Genetics; HOSCO, Saint Camille Hospital of Ouagadougou; USTA, University Saint Thomas d’Aquin.
REFERENCES
|
Bakker E, Jennekens FG, De Visser M, Wintzen AR (1997). Duchenne and Becker muscular dystrophies In: Diagnostic criteria for neuromuscular disorders, pp. 1-4.
|
|
|
|
Buddhe S, Cripe L, Friedland-Little J, Kertesz N, Eghtesady P, Finder J, Hor K, Judge DP, Kinnett K, McNally EM, Raman S, Thompson WR, Wagner KR, Olson AK (2018). Cardiac management of the patient with duchenne muscular dystrophy. Pediatrics 142(2):S72-S81.
Crossref
|
|
|
|
|
Bushby KM, Gardner-Medwin D, Nicholson LV, Johnson MA, Haggerty ID, Cleghorn NJ, Harris JB, Bhattacharya SS (1993). The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. II. Correlation of phenotype with genetic and protein abnormalities. Journal of Neurology 240(2):105-112.
Crossref
|
|
|
|
|
Campbell KP (1995). Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80(5):675-679.
Crossref
|
|
|
|
|
Cripe LH, Tobias JD (2013). Cardiac considerations in the operative management of the patient with Duchenne or Becker muscular dystrophy. Pediatric Anesthesia 23(9):777-784.
Crossref
|
|
|
|
|
Deburgrave N, Daoud F, Llense S, Barbot JC, Recan D, Peccate C, Burghes AH, Beroud C, Garcia L, Kaplan JC, Chelly J, Leturcq F (2007). Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Human Mutation 28(2):183-195.
Crossref
|
|
|
|
|
Gospe SM, Lazaro RP, Lava NS, Grootscholten PM, Scott MO, Fischbeck KH (1989). Familial X-linked myalgia and cramps: a nonprogressive myopathy associated with a deletion in the dystrophin gene. Neurology 39(10):1277-1280.
Crossref
|
|
|
|
|
Kwiatkowska J, Meyer-Szary J, Bazgier M, Fijałkowska J, Wierzba J, Glińska A, Dorniak K (2020). Left ventricular volumes and function affected by myocardial fibrosis in patients with Duchenne and Becker muscular dystrophies: a preliminary magnetic resonance study. Kardiologia Polska, pp. 1-10.
Crossref
|
|
|
|
|
Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, Angelini C, Dalla Volta S (1996). Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy. Circulation 94(12):3168-3175.
Crossref
|
|
|
|
|
Mori-Yoshimura M, Mitsuhashi S, Nakamura H, Komaki H, Goto K, Yonemoto N, Takeuchi F, Hayashi YK, Murata M, Takahashi Y, Nishino I, Takeda S, Kimura E (2018). Characteristics of Japanese Patients with Becker Muscular Dystrophy and Intermediate Muscular Dystrophy in a Japanese National Registry of Muscular Dystrophy (Remudy): Heterogeneity and Clinical Variation. Journal of Neuromuscular Diseases 5(2):193-203.
Crossref
|
|
|
|
|
Mornet D, Rivier F (2017). What future for dystrophin? Les cahiers de myologie (15):17-21.
Crossref
|
|
|
|
|
Muntoni F, Torelli S, Ferlini A (2003). Dystrophin and mutations: one gene, several proteins, multiple phenotypes. The Lancet Neurology 2(12):731-740.
Crossref
|
|
|
|
|
Nakamura A (2015). X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals (Basel) 8(2):303-320.
Crossref
|
|
|
|
|
Ortez C, Natera de Benito D, Carrera Garcia L, Exposito J, Nolasco G, Nascimento A (2019). Advances in the treatment of Duchenne muscular dystrophy. Medicina 79(3):77-81.
|
|
|
|
|
Péréon Y, Mercier S, Magot A (2015). Duchenne muscular dystrophy pathophysiology. Archives de pédiatrie. 22(12Suppl1):12S18-23
Crossref
|
|
|
|
|
Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Academy of Sciences of the United States of America 90(8):3710-3714.
Crossref
|
|
|
|
|
Rahimov F, Kunkel LM (2013). The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. Journal of Cell Biology 201(4):499-510.
Crossref
|
|
|
|
|
Serrano AL, Munoz-Canoves P (2017). Fibrosis development in early-onset muscular dystrophies: Mechanisms and translational implications. Seminars in cell and developmental biology 64:181-190.
Crossref
|
|
|
|
|
Straathof CS, Van Heusden D, Ippel PF, Post JG, Voermans NC, De Visser M, Brusse E, Van Den Bergen JC, Van Der Kooi AJ, Verschuuren JJ, Ginjaar HB (2015). Diagnosis of becker muscular dystrophy: Results of Re-analysis of DNA samples. Muscle and Nerve 53(1):44-48.
Crossref
|
|