Full Length Research Paper

ABSTRACT
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2), associated with Corona Virus Disease 2019 (COVID-19) coined by World Health Organization, belongs to single stranded RNA viruses (ssRNA Viruses) under Betacoronaviruses. The virus’ molecular dynamics are necessary in the wake of human-human transmissions globally with mortality cases on the rise, thusly the race for a vaccine. As the viral genome expresses more human-biased mutations, the coronavirus disease 2019 continues to infect people in their millions, with the available detection kits limiting the numbers detected out of the population. Understanding the molecular basis of the virus through bioinformatics would speed up the viral diagnostics, management and vaccine generation. Currently, the scientific community seeks to give varied perspectives of what is known of the virus at a cellular level. The knowledge is scattered and requires a consolidated flow on thematic understanding in order to ensue further build up towards curbing the disease. The structure and function of the virus, genome and revealed mutations are critical in directing the SARS-CoV-2 virus and disease understanding. Here, we analyze and review published knowledge on the virus in relation to the molecular specs and evolutionary relationships of the virus.
Key words: SARS-CoV-2 infection, COVID-19, RNA viruses.
INTRODUCTION
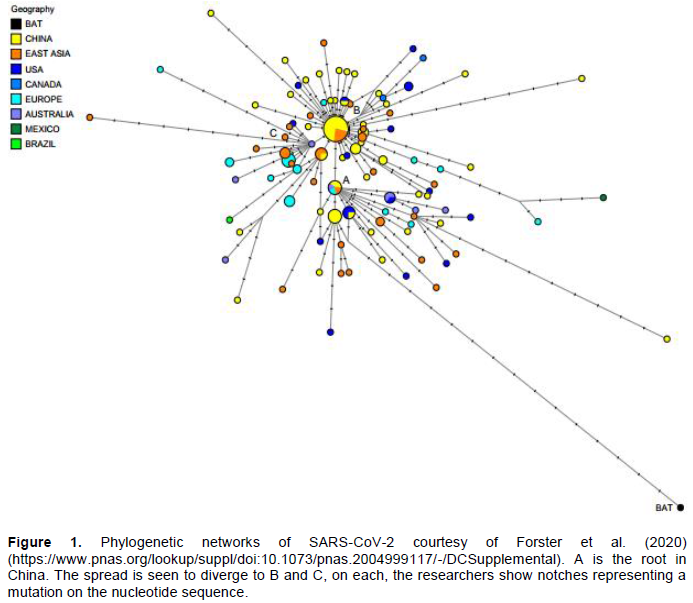
METHODOLOGY
RESULTS

DISCUSSION
CONCLUSION
CONFLICT OF INTERESTS
The authors have not declared any conflict of interests.
REFERENCES
|
Alagaili AN, Briese T, Mishra N, Kapoor V, Sameroff SC, de Wit E, Munster VJ, Hensley LE, Zalmout IS, Kapoor A, Epstein JH (2014). Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio 5(4):e01482-14. |
|
|
Asim B, Uttaran B, Alok Kumar C, Devendra NT, Hasina B, Shanta D (2020). Emergence of Novel Coronavirus and COVID-19: Whether to stay or die out? Critical Reviews in Microbiology doi10.1080/1040841X.2020.1739001. |
|
|
Chan JFW, Kok KH, Zhu Z, Chu H, To KK-W, Yuan S, Yuen K-Y (2020). Genomic characterization of the 2019 novel humanâ€pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes and Infections 9:221â€236. |
|
|
Chan JFW, Lau SKP, To KKW, Cheng VCC, Woo PCY, Yuen K (2015). Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARSâ€like disease. Clinical Microbiology Reviews 28:465â€522. |
|
|
Changtai W, Zhongping L, Zixiang C, Xin H, Mengyuan X, Tengfei H, Zhenhua Z (2020). The establishment of reference sequence for SARSâ€CoVâ€2 and variation analysis. Journal of Medical Virology 92:667-674. |
|
|
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Yu T (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 395:507â€513. |
|
|
Chu DKW, Poon LLM, Gomaa MM, Shehata MM, Perera RA, Zeid DA, El Rifay AS, Siu LY, Guan Y, Webby RJ, Ali MA (2014). MERS coronaviruses in dromedary camels, Egypt. Emerging Infectious Diseases 20:1049â€1053. |
|
|
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Research 176:104742. |
|
|
Cui J, Li F, Shi ZL (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology 17(3):181-192. |
|
|
Cyranoski D (2020). Did pangolins spread the China coronavirus to people?. Nature. |
|
|
Eickmann M (2003). Phylogeny of the SARS Coronavirus. Science 302(5650):1504b-11505. |
|
|
Felsenstein J (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39:783-791. |
|
|
Forster P, Lucy F, Colin R, Michael F (2020). Phylogenetic network analysis of SARS-CoV-2 genomes. doi:10.1073/pnas.2004999117. |
|
|
Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA (2020). Severe acute respiratory syndrome-related coronavirus: The species and its viruses - A statement of the Coronavirus Study Group. |
|
|
Grubaugh ND, Petrone ME, Holmes EC (2020). We shouldn't worry when a virus mutates during disease outbreaks. Nature Microbiology 5:529-530. |
|
|
Guan WJ, Ni Z, Hu Y, Liang W, Ou CQ, He Jx, Liu L, Shan H, Lei CL, Hui DSC, Du B, Li LJ, Zeng G, Kowk-Yung Y, Ru-chong C, Chun-li T, Tao W, Ping-yan C, Jie X, Shi-yue L, Jin-lin W, Zi-jing L, Yi-xiang P, Li W, Yong L, Ya-hua H, Peng P, Jian-ming W, Ji-yang L, Zhong C, Gang L, Zhi-jian Z, Shao-qin Q, Jie L, Chang-jiang Y, Shao-yong Z, Nan-shan Z (2019). Clinical characteristics of 2019 novel coronavirus infection in China. medRxiv. 2020:20020974. |
|
|
Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM (2003). Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 302(5643):276-278. |
|
|
Hall TA (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41:95-98. |
|
|
Huang C, Wang Y, Li X, Ren, L., Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497â€506. |
|
|
Hurst KR, Koetzner CA, Masters PS (2013). Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. Journal of Virology 87(16):9159-9172. |
|
|
Koyama T, Platt D, Parida L (2020). Variant analysis of COVID-19 genomes. Bull. World Health Organ. https://doi.org/10.2471/BLT.20.253591. |
|
|
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution 35:1547-1549. |
|
|
Lam TTY, Shum MHH, Zhu HC, Tong YG, Ni XB, Liao YS, Wei W, Cheung WYM, Li WJ, Li LF, Leung GM, Holmes EC, Hu YL, Guan Y (2020). Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. bioRxiv. |
|
|
Lau SKP, Woo PCY, Li KSM, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY (2005). Severe acute respiratory syndrome coronavirusâ€like virus in Chinese horseshoe bats. Proceedings of the National Academy of Sciences of the United States of America 102:14040â€14045. |
|
|
Lefkowitz EJ, Dempsey DM, Hendrickson RC, Orton RJ, Siddell SG, Smith DB (2018). Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Research 46(D1):D708-D717. |
|
|
Li W, Shi Z, Yu M, Wuze R, Craig S, Jonathan HE, Hanzhong W, Gary C, Zhihong H, Huajun Z, Jianhong Z, Jennifer M, Hume F, Peter D, Bryan TE, Shuyi Z, Lin-Fa W (2005). Bats are natural reservoirs of SARSâ€like coronaviruses. Science 310:676â€679. |
|
|
Li X, Junjie Za, Qiang Z, Qing N, Yi L, Brian TF, Antoine C (2020a). Evolutionary history, potential intermediate animal host, and crossâ€species analyses of SARSâ€CoVâ€2. Journal of Medical Virology. |
|
|
Li X, Wang W, Zhao X, Zai J, Zhao Q, Li Y, Chaillon A (2020b). Transmission dynamics and evolutionary history of 2019â€nCoV. Journal of Medical Virology. |
|
|
Li X, Zai J, Wang X, Li Y (2020c). Potential of large 'first generation' humanâ€toâ€human transmission of 2019â€nCoV. Journal of Medical Virology 92:448â€454. |
|
|
Lu R,Zhao X, Li J,Niu P, Yang B, Honglong W, Wenling W, Hao S, Baoying H, Na Z, Yuhai B, Xuejun M, Faxian Z, Liang W, Tao H, Hong Z, Zhenhong H, Weimin Z, Li Z, Jing C, Yao M, Ji W, Yang L, Jianying Y, Zhihao X, Jinmin M, William JL, Dayan W, Wenbo X, Edward CH, George FG,Guizhen W, Weijun C, Weifeng S, Wenjie T (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. The Lancet 395(10224):565-574. |
|
|
Marra MA, Jones SJM, Astell CR, Holt RA, Brooks-Wilson A, Yaron SNB, Jaswinder K, Jennifer KA, Sarah AB1, Susanna YC, Alison C1, Shaun MC, Doug F, Noreen G, Obi LG, Stephen RL, Michael M, Helen M, Stephen BM, Pawan KP, Anca SP, Gordon AR, Jacqueline ES, Asim S, Duane ES, Jeff MS, George SY, Francis P, Anton A, Harvey A, Nathalie B, Kathy B, Timothy FB, Donnie B, Martin C, Michael D, Lisa F, Ramon F, Michael G, Michael G, Allen G, Steven J, Heinz F, Adrienne M, Amin K, Yan L, Susan N, Ute S, Graham AT, Shaun T, Robert V, Diane W, Brynn W, Robert CB, Mel K, Martin P, Danuta MS, Chris U, Rachel LR (2003). The genome sequence of the SARS-associated coronavirus. Science 300(5624):1399-1404. |
|
|
Müller MA, Corman VM, Jores J, Meyer B, Younan M, Liljander A, Bosch BJ, Lattwein E, Hilali M, Musa BE, Bornstein S, Drosten C (2014). MERS coronavirus neutralizing antibodies in camels, Eastern Africa, 1983â€1997. Emerging Infectious Diseases 20:2093â€2095. |
|
|
NCBI Repository (National Center for Biotechnology Information, U.S). National Library of Medicine 8600 Rockville Pike, Bethesda MD, 20894 USA. Available at https://www.ncbi.nlm.nih.gov. |
|
|
Niemeyer D, Mösbauer K, Klein EM, Andrea S, Robert CM, Anna MM, Ronald D,Susan CB, Christian D, Marcel AM (2018). The papain-like protease determines a virulence trait that varies among members of the SARS-coronavirus species. PLoS Pathology 14(9):e1007296. |
|
|
Oostra M, de Haan CA, Rottier PJ (2007). The 29-nucleotide deletion present in human but not in animal severe acute respiratory syndrome coronaviruses disrupts the functional expression of open reading frame 8. Journal of Virology 81:13876-13888. |
|
|
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen M-H, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TCT, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Günther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ (2003). Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300(5624):1394-1399. |
|
|
Rozhgar AK, Muhamad S, Mehmet O (2020). Genomic characterization of a novel SARS-CoV-2. |
|
|
Saitou N, Nei M (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4:406-425. |
|
|
Shi CS, Nabar NR, Huang NN, Kehrl JH (2019). SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discovery 5;101. |
|
|
Song HD, Tu CC, Zhang GW, Sheng-Yue W, Kui Z, Lian-Cheng L, Qiu-Xia C, Yu-Wei G, Hui-Qiong Z, Hua X, Hua-Jun Z, Shur-Wern WC, Feng C, Chun-Ming P, Hua X, Sai-Juan C, Hui-Ming L, Duan-Hua Z, Yu-Fei L, Jian-Feng H, Peng-Zhe Q, Ling-Hui L, YQR, Wen-Jia L, Ye-Dong Y, Larry A, Ming W, Rui-Heng X, Xin-Wei W, Huan-Ying Z, Jin-Ding C, Guodong L, Yang G, Ming L, Ling F, Li-Yun J, Hui L, Fang C, Biao D, Li-Juan H, Jin-Yan L, Suxiang T, Xiangang K, Lin D, Pei H, Hua T, Andrea B, Xiao-Jing Y, Ottavia S, Zong-Ming G, Hai-Yan P, Wei-Zhong H, Jean-Claude M, Arnaud F, Antoine D, Neri N, Yi-Xue L Chung-I W, Guo-Ping Z (2005). Crossâ€host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proceedings of the National Academy of Sciences of the United States of America 102:2430â€2435. |
|
|
Su S, Wong G, Shi W, Liu J, Lai ACK, Zhou J, Liu W, Bi Y, Gao GF (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiology 24:490â€502. |
|
|
Wang M, Yan M, Xu H,Liang W, Kan B, Zheng B, Chen H, Zheng H, Xu Y, Zhang E, Wang H, Ye J, Li G, Li M, Cui Z, Liu Y-F, Guo R-T, Liu X-N, Zhan L-H, Zhou D-H, Zhao A, Hai R, Yu D, Guan Y, Xu J (2005). SARSâ€CoV infection in a restaurant from palm civet. Emerging Infectious Diseases 11:1860â€1865. |
|
|
Woo CYP, Wang M, Lau SKP, Huifang X, Rosana WSP, Rongtong G, Beatrice HL W, Kai G, Hoi-Wah T, Yi H, Kenneth SML, Carol SFL, Kwok-Hung C, Bo-Jian Z, Kwok-Yung Y (2007). Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. Journal of Virology 81(4):1574-1585. |
|
|
World Health Organization (WHO) (2020) Novel Coronavirus (COVID-19) Situational Reports. Available at View (30th April 2020, date last accessed). |
|
|
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260-1263. |
|
|
Wu F, Zhao S, Bin Y, Yan-Mei C, Wen W, Zhi-Gang S, Yi H, Zhao-Wu T, Jun-Hua T, Yuan-Yuan P, Ming-Li Y, Yu-Ling Z, Fa-Hui D, Yi L, Qi-Min W, Jiao-Jiao Z, Lin X, Edward CH, Yong-Zhen Z (2020). A new coronavirus associated with human respiratory disease in China. Nature. |
|
|
Xiao K, Zhai J, Yaoyu F, Niu Z, Xu Z, Jie-Jian Z, Na L, Yaqiong G, Xiaobing L, Xuejuan S, Zhipeng Z, Fanfan S, Wanyi H, Yu L, Ziding Z, Rui-Ai C, Ya-Jiang W, Shi-Ming P, Mian H, Wei-Jun X, Qin-Hui C, Fang-Hui H, Yahong L, Wu C, Lihua X, Yongyi S (2020). Isolation and characterization of 2019-nCoV-like coronavirus from malayan pangolins. bioRxiv. |
|
|
Zhang T, Wu QF, Zhang ZG (2020). Pangolin homology associated with 2019-nCoV. bioRxiv. |
|
|
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao X, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. |
|
|
Zhu N, Zhang D, Wenling W, Xingwang L, Bo Y, Jingdong S, Xiang Z, Baoying H, Weifeng S, Roujian L,Peihua N, Faxian Z, Xuejun M, Dayan W, Wenbo X, Guizhen W, George FG, Wenjie T (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine 382(8):727-733. |
|
|
Zuckerkandl E, Pauling L (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by Bryson V, Vogel HJ. Academic Press, New York. pp. 97-166. |
|
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


