Full Length Research Paper

ABSTRACT
Following the screening of several plant species from an inventory of common medicinal plants from South Africa for medicinal properties, Cissampelos capensis was selected for further investigation due to its interesting and useful ethnomedicinal properties. This study attempts to relate specific constituents present in this plant with its widespread ethnomedicinal uses. Six compounds were isolated and their structures were unambiguously established by spectroscopic methods. The compounds are: 5,6-dehydro-4,5-dihydroxy-1,3,6-trimethoxy-17-methylmorphinan-7-one (1); 1,2-methylenedioxy-3-hydroxy -9,10-dimethoxyaporphine (2); 5,6-didehydro-4-hydroxy-3,6-dimethoxy-17-methylmorphinan-7-one (3); 3,7,8,3'-tetramethoxy- 6 - C-methyl- 5,4'-dihydroxyflavone (6 -C-methylquercetin 3, 3',7, 8 -tetramethyl ether) (4); 5, 7, 8 -trihydroxy-2′, 5′-dimethoxy-3′,4′- methylenedioxyisoflavanone (5); 3 -methoxy-6 -C-methyl-3',4',5,7,8 -pentahydroxyflavone (6 -C- methylquercetin -3-methyl ether) (6). Five of the isolated compounds, (viz., 1,2,4,5 and 6) have, to our knowledge, not been reported previously. The crude fractions and isolates were tested for cytotoxicity using the brine shrimp lethality test and for antimicrobial properties using nine microbes, including three Gram –ve, three Gram +ve bacteria and three fungi. The Gram-negative bacteria were Pseudomonas aeruginosa (NCTC 10332), Proteus vulgaris (NCTC 4175) and Escherichia coli Sero type 1 (NCTC 09001), while the Gram-positive bacteria were Bacillus subtilis (NCTC 8236), Staphylococcus aureus (NCTC 13134) and Bacillus licheniformis (NCTC 01097). The Fungal species used were Candida albicans (ATCC 90028), Candida eropiralis (ATCC 750) and Aspergillus niger (ATCC 10578). The n-Hex fractions were not active while the highest activities were found in the methanolic extracts. The total tertiary alkaloid fraction (TTA) showed the highest activity against the Bacillus substillis. Compounds 1, 2 and 5 appear to be the most promising with regards to the prospects of drug development.
Key words: Phytochemistry, Cissampelos capensis, antimicrobial, cytotoxicity, isoflavanone, aporphine, morphinandienone.
INTRODUCTION
Medicinal plants, in a variety of forms have been recorded to cure, manage and control conditions in people for at least four thousand years, and it may be assumed that this practice extends even longer (Christophersen et al., 1991). The World Health Organisation (WHO) has recognized this and has accepted it as an essential building block for primary health care (Akerele, 1988; WHO, 2005). The vibrant healing power of C. capensis has been recognized in recent times (Van and Gericke, 2000; Iwu, 1993; WHO, 2002), and especially by traditional healers in South Africa, who are known by the Zulu people as inyangas, herbalists, isangomas or diviners while they are also known as amaxhwele, amagqira (Xhosa), ngaka (Sotho),nanga, mungome or maine (Vhavenda) (Van, et al., 1997; Mander, 1998; De Wet and Van, 2008; Van, 2008; Van et al., 2008).
The plant is one out of 520 species in 75 genera (Van et al., 1997), which belongs to the family of menispermaceae. It is widely distributed in the sandy slopes and scrublands of the Northern, Western and Eastern Cape Provinces of South Africa and northwards into Namibia. It is called by different local name such as Dawidjiewortel (Afrikaans) and Mayisake (Xhosa). Due to its richness in bisbenzylisoquinoline alkaloids, this family is used worldwide in traditional medicine to treat a variety of ailments especially cancer (Barbosa et al., 2000; DeWet et al., 2004, 2005; DeWet, 2006; Watt and Breyer-Brandwijk, 1962; Smith, 1966; Rood, 1994; Cillie, 1992; Dykman, 1891; Iwu, 1993). It is a woody dioecious perennial climbing vine with a twining stem, shrublet without tendrils and supports itself by twining around the stems of other plants, fence or walls. The leaves are rounded, bright green, almost without hairs. The flowers, which usually sprout between February and May, are axillary with velvety-hairy and greenish in colour. The fruit is inedible, dark, grape-sized berries. The root may be up to 2.5cm in diameter with a grey-brown bark (Botha, 1980). The plant is usually confused with Zehneria scabra (Cucurbitaceae), which in some parts is known by the same local name (Smith, 1966).
The latter resembles a cucumber and can be distin-guished by the spirally coiled tendrils. Nearly all parts of the plant are used; from the whole vine, seed, bark, leaf to the root. It is traditionally used in South Africa to treat a variety of ailments such as dysentery, menstrual prob-lems, prevention of miscarriage, cholera, colic, snakebite, measles, fever, diabetes, tuberculosis, stomach and skin cancers, etc (Watt and Breyer-Brandwijk, 1962; Rood, 1994; Van Wyk and Gericke, 2000; VanWyk et al., 1997; De Wet and Van Wyk, 2008; Barbosa- Filho et al., 2000; De Wet et al., 2004, 2005). In 1962, it was reported that C. pareira demonstrated anti-inflammatory, smooth muscle relaxant, antispasmodic, diuretic effect and uterine relaxant actions in various laboratory animals and this was further documented by Pillay et al. (2008) and Taylor (2005). Ssegawa and Kasenene, (2007) described its antiulcerous actions and mild hypoglycemic activity. Further studies have also shown that the roots described its antiulcerous actions and shown that the roots of cissampelos species have anticonvulsant activity, antioxidant properties, antimicrobial activity, antihypertensive, antimalarial effects, antispasmodic and antitumour properties (Dic. Nat. prod., 1996,2006; Bruneton, 1995; Wu, 2007; Amresh et al., 2007; Amresh et al., 2004; Gessler et al., 1995; Ramirez et al., 2003; Sanchez et al., 2001; Taylor, 2005; Graham et al., 2000; Hamill et al., 2003). The antinociceptive and antiarthritic activity of Cissampelos pareira roots were also highlighted by Amresh et al., (2007) while the antifertility activity and anti-inflammatory activity were described by Ganguly et al. (2007) and Amresh et al. (2007) respectively. The Khoi-San and the Cape Dutch recognised the importance of this plant in Cape herbal medicine as one of the major distinct plants used for the treatment of most diseases (Van Wyk, 2008), while during the evaluation of some South African medicinal plants for antimalarial properties, it was discovered that C. capensis possesses antiplasmodial activity (Pillay et al., 2008). The medicinal values of Cissampelos species in the Sango bay area in Southern Uganda was also recorded by Ssegawa and Kasenene (2007).
Phytochemical screening showed the presence of alkaloids, saponins, flavonoids, phenolics and tannins while cardiac glycosides and anthraquinones were not detected in the different extracts (Babajide et al, 2010). A large number of biologically active alkaloids have been isolated from several Cissampelos species among these were cissampareine and magnoflorine, (Jittra et al., 2005; Freitas et al., 1995). Other alkaloids such as cissaglaberrimine and oxobuxifoline were isolated from Cyathea glaberrima (Barbosa-filho et al., 1997) while Milonine, an 8, 14-dihydromorphinandienone alkaloid was also isolated from the dried leaves of Cissampelos sympodialis (Freitas et al., 1995). Similarly, Sloan et al. (2007) showed the isolation of two known aporphine alka-loids, (S)-dicentrine and (S)-neolitsine from methanolic extract of C. capensis, which were found to be potent anthelmintics. To the best of our knowledge no flavonoidal compounds isolated from C. capensis has been published.
The present study involves a more intense investigation of the constituents present in C. capensis as well as their biological activities.
MATERIALS AND METHODS
General experimental proceduresAll laboratory grade solvents were distilled prior to use. Spectroscopic grade solvents were used as such. Adsorption column chromatography was performed using Merck Silica gel 60 H (0.040 -0.063 mm particle size, Merck).
Unless otherwise specified, a column with internal diameter of 25 mm was used and volumes of 10 ml were collected in Pyrex test tubes. Size-exclusion column chromatography was performed using Sephadex LH-20 (Sigma) pre-swollen in the specified solvent before loading of sample.
Preparative Thin Layer Chromatography (TLC) was performed using Merck Silica gel 60 PF254 on glass plates (20 cm x 20 cm) and with a thickness of 0.5 mm. Analytical TLC was conducted on normal-phase Merck Silica gel 60 PF254 on precoated aluminium plates. Separated compounds on TLC and Preparative Thin Layer Chromatography (PTLC) plates were visualized under Ultraviolet (UV) light at (254 and 366 nm), and spraying of the plates where required were carried out using 2% vanillin in H2SO4 followed by heating at 110°C for 2 to 4 mins. Quercetin, rutin and catechin were used as reference standards for flavonoids, gallic acid for tannins and oleanolic acid for essential oils. In the case of the Alkaloids, the dragendorff spraying reagent was used for visualization while quinine and berberine were used as standards.
Reversed phase TLC (RPTLC) was performed using pre-coated plates (Merck, RP-18. F254 2, 0.25 mm thickness) and spots were detected as described above. Rf values were determined at room temperature using different solvent systems as applicable. Melting points were determined on a Fisher – John’s melting point apparatus (Fisher-Scientific). All melting points are uncorrected. Mass spectra were obtained using JEOL JMS-AX505HA double-focusing probe at 70 eV while UV spectra with a Unicam UV 4 -100 UV/Vis recording spectrophotometer. The spectra were recorded over the range of 200 to 550 nm, at a concentration of 0.02 mg/ml in spectroscopic grade methanol. All infra-red (IR) spectra were recorded on a Perkin Elmer universal ATR Spectrum 100 series FT-IR spectrometer while Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AMX-400 (400 MHz for 1H and 100 MHz for 13C) spectrometer, using a triple resonance probe head with self-shielded gradient coils and a Bruker Z-gradient accessory delivery squared gradients. The chemical shifts are expressed in ppm relative to tetramethylsilane (TMS).
Plant
The aerial shoot and root of C. capensis were collected in May 2007 from the University of the Western Cape (UWC) Cape Nature Conservation Reserve, Bellville, Cape Town, South Africa, authenticated by a plant systematist and a voucher specimen [number Weitz 1056(UWC)] deposited in the hebarium of the University of the Western Cape. Preparation of the extracts was performed as described by Babajide et al. (2010).
Extraction and isolation
Each of the extracts that is, hexane (Hex), dichloromethane (DCM), ethyl acetate (EtOAc), methanol (MeOH), TTA and residual aqueous fraction (BTA) were separately evaporated under reduced pressure at 40°C while aqueous extracts were concentrated by freeze-drying. The preliminary phytochemical screening (Wagner and Bladt, 2001) was carried out for detection of classes of secondary plant metabolites as shown in Babajide et al. (2010)
Fractionation of the TTA
The TTA (1.6g) was adsorbed on silica gel and the constituents were separated by column gradient chromatography (CGC) using an increasing gradient of Hex and CHCl3 and finally MeOH was employed. The fractions collected were analyzed by TLC using Hex– CHCl3 – MeOH (1: 5 : 1). Tubes showing similar TLC characteristics were bulked together and concentrated. Seven fractions coded A to G were obtained and the bioactivities of each were monitored using the brine shrimp cytotoxicity test. Fraction E (0.30g) with LC50 value of 2.4050 was selected for further fractionation. Fraction E (0.25 g) was rechromatographed using CGC to afford five fractions: TI to TV and fractions TIII (0.122g) with an LC50 value of 3.0226 and TIV (0.047g) with an LC50 value of 0.8655 were further investigated. Repeated preparative TLC of TIII gave compound (1) a brownish yellow powder (0.058 g) and that of TIV afforded compound (2) as a brown amorphous solid (0.021g).
Extraction and fractionation of the aerial shoot
The aerial shoot (1 kg) was sequentially extracted to afford the hexane (17.35g, 1.74%), dichloromethane (26.33g, 2.63%), ethyl acetate (32.84g, 3.28%), methanol (56.24g, 5.62%) and water extracts (25.71g, 2.57%). The methanol extract (25.6g) was chromatographed using same method as described for TTA. Ten fractions coded A to J were obtained and fraction F (1.96g) with an LC50 value of 1.2332 was selected for further fractionation which afforded five fractions viz., FI to FV of which FII (0.520g) with LC50 of 66.73, FIII (0.211g) with LC50 of 3.442, FIV (0.115g) with LC50 of 17.56 and FV (0.106g) with LC50 of 45.46 were further investigated. Fraction FII (0.5 g) was chromatographed using CGC and was further purified using sephadex LH-20 and repeated preparative TLC. This gave a golden brown powder (0.04g) of compound (3). Fraction FIII (0.20 g) was also rechromatographed and further subjected to repeated preparative TLC to afford a brownish yellow powder which was recrystallized from EtOH to give a further quantity of compound (1) (a brownish yellow powder, 0.028 g) while fraction FIV (0.10 g) after preparative TLC afforded (4) as a yellow amorphous powder (0.026 g) and fraction FV (0.10 g) gave compound (5) as a yellow powder (0.024g).
Extraction and fractionation of the root
The same extraction procedure was repeated for the dried root (1 kg). This afforded a hexane (7.38g, 0.74%), dichloromethane (12.65g, 1.27%), ethyl acetate (27.19g, 2.72%), methanol (53.05g, 5.31%) and water extract (26.44g, 2.64%). The methanol extract (30.0g) was chromatographed as described for the aerial shoot. Out of the five fractions collected, fractions M (5.68g) with LC50 value of 5.2375 and N (3.67g) with LC50 of 19.296 were selected for further fractionation. The fractionation of M (5.0 g) by repeated CGC and monitoring of their LC50 values resulted more in compounds of (1) (0.078g), (2) (0.065g) and (5) (0.088g). Similarly, fraction N (3.2 g) was rechromatographed and accorded more of compound (4) (0.068g) and a new compound viz., (6) (0.135g) a creamy yellow solid was obtained from subfractions NIII (0.73 g) with LC50 of 76.112.
Dihydromorphinandienone alkaloid (1)
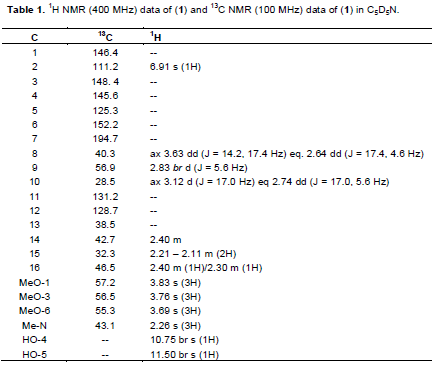
A reddish yellow powder with m.p. 85 to 87°C; UV λmax nm (log ε): 222, 260, 304; IR nmax cm-1 pronounced peaks: 3485, 2940, 1685, 1615, 1500; EIMS: molecular ion peak (M)+. m/z 375.0216 ( calculated for C20H25O6N: 375.1682). The 1H NMR and 13C NMR see Table 1
9, 10 – Dimethoxyaporphine alkaloid (2)
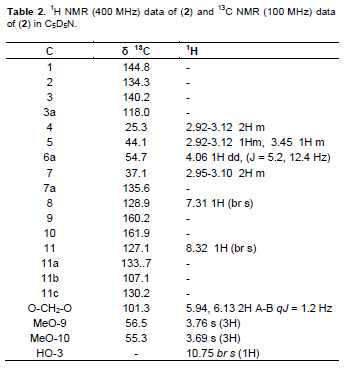
A reddish-brown fluffy powder with m.p. 221 – 223°C; UV λmax nm(log ε): 225, 243, 277, 300 (shoulder). IR nmax cm-1 pronounced peaks: 3420, 3050, 2925, 1636, 1458, 1387, 1060. EIMS: molecular ion peak (M)+. m/z 341.0112. (calculated for C19H19O5N: 341.1263). For the 1H NMR and 13C NMR (Table 2).
8,14-Dihydromorphinandienone alkaloid (3)
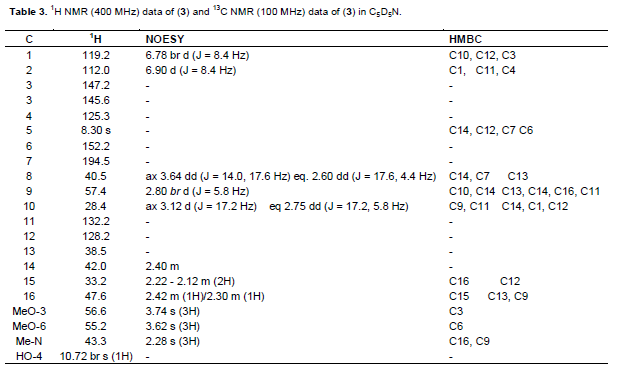
A golden brown powder with m.p 77 to 80°C. UV λmax nm (log ε): 226, 261,302; IR (ν cm-1) pronounced peaks: 3517; 2952, 1685, 1616, and 1510. EIMS: the molecular ion base peak (M)+ m/z 329.2133 (calculated for C19H23O4N: 329.1627). The 1H NMR and 13C NMR (Table 3)
6 -C-methylquercetin -3, 3',7, 8 -tetramethyl ether (4)
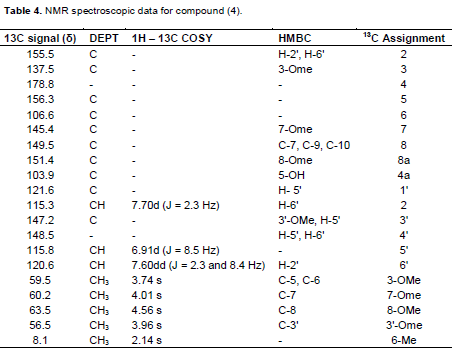
A yellow amorphous powder with m.p. 234 to 236°C. UV λmax nm (log ε): 255 (4.64), 357 (4.62); +NaOAc: 271, 365; +NaOH: 276, 343, 410. +AlCl3: 279, 440. IR (ν cm-1) pronounced peaks: 3401, 2932, 1653, 1612, 1556 EIMS: molecular ion peak (M)+. m/z 388.0103. ( calculated for C20H20O8 : 388.1158 ). 1H NMR: δ 2.14 (3H, s, Me-6), 3.74 (3H, s, OMe-3), 3.96 (3H, s, OMe-3'), 4.01 (3H, s, OMe-7); 4.56 (3H, s, OMe-8), 6.91 (1H, d, J= 8.5 Hz, H-5'), 7.60 (1H, dd, J = 2.3, 8.4 Hz, H-6'), 7.70 (1H, d, J= 2.3 Hz, H-2'), 10.90 (1H, s, 4'-OH), 12.92 (1H, s, 5-OH). 13C NMR (Table 4)
Methylenedioxyisoflavanone (5)
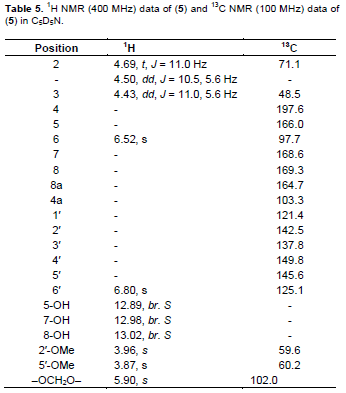
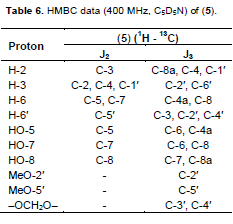
A yellow waxy powder with m.p. 185 to 187°C; UV lmax nm (log ε): 214(4.10), 218 (4.12), 288 (4.00), 333 (sh) (3.25). IR nmax cm−1 pronounced peaks at : 3421, 2914, 1634, 1472, 1386, 1358, 1257, 1224, 1161, 1100, 1068, 925; EIMS: molecular ion peak (M)+. m/z 378.0625 (calculated for C18H18O9, 378.0623). 1H NMR, 13C NMR and HMBC are as shown in Tables 5 and 6 respectively.
6-C-Methylquercetin 3-methyl ether (6)
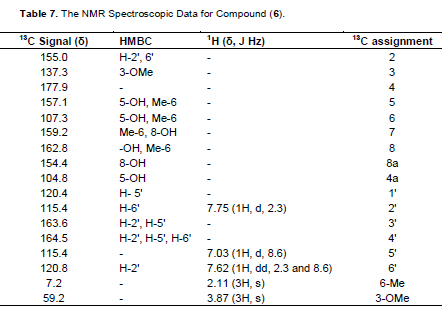
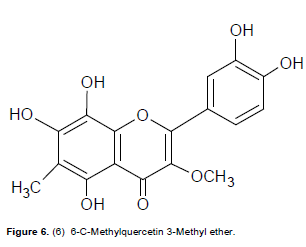
A creamy yellow powder with m.p. 217 to 219°C. UV λmax nm (logε): 260 (4.62), 351 (4.60); +NaOAc: 260, 355; +NaOH: 270, 405; +A1C13: 277, 435. IR (ν cm-1) pronounced peaks: 3500 – 3400, 1652, 1613, 1568, 1556 EIMS: molecular ion peak (M)+. m/z 346.0752 ( calculated for C17H14O8 : 346.0688). 1H NMR: δ 2.11 (3H, s, Me-6), 3.87 (3H, s, OMe-3), 7.03 (1H, d, J=8.6Hz, H-5'), 7.62 (1H, dd, J= 2.3, 8.6 Hz, H-6'), 7.75 (1H, d, J= 2.3Hz, H-2'), 10.23 (1H, s, 4'-OH), 11.15 (1H, s, 3’-OH).12.05 (1H, s, 8-OH), 12.90 (1H, s, 7-OH), 13.05 (1H, s, 5-OH). 13C NMR: see Table 7.
Brine shrimp lethality biossay
The brine shrimp lethality biossay was carried out as previously described ( Babajide et al., 2008, 2010) where about 2 g of the Brine shrimp eggs (Artemia salina Leach) were hatched in 2 L of sea water using a large plastic case as an artificially partitioned dam. The number of dead nauplii were counted and recorded (lethality data) after 24 h. The numbers of dead nauplii were used for calculating the LC50 at 95% confidence limit by the Finney Probit analysis program. LC50 values greater than 800 ppm or in that range were considered inactive.
Antimicrobial activity
The organisms used in the screening tests were as follows: The gram-negative bacteria were P. aeruginosa (NCTC 10332), P. vulgaris (NCTC 4175) and E. coli sero type 1 (NCTC 09001), while the gram-positive bacteria were B. subtilis (NCTC 8236), S. aureus (NCTC 13134) and B. licheniformis (NCTC 01097). These species are considered as the most important pathogens (NCCLS, 1990). Fungal species used were C. albicans (ATCC 90028), C. eropiralis (ATCC 750) and A. Niger (ATCC 10578). Cultures were grown in sabouraud dextrose (SD) broth at 37°C and maintained on SD agar at 4°C. A colony of each bacterial strain was suspended in 1 ml of Mueller–Hinton broth and incubated for 18 h at 37°C. A subculture was again made after 6 h. The subculture was diluted 1/50 in the same broth before use. A disc diffusion assay was used to determine the inhibition of bacterial growth by the plant extracts, fractions, sub fractions and isolates (Rasoanaivo et al.,1993). Plant extracts, fractions and isolates were dissolved in appropriate solvents at a concentration of 100 mg/ml for extracts and 10 mg/ml for isolates. 20 μl were dispensed on a 9 mm sterile paper disc (Munktell/Lasec, Numb. FLAS3526009). Amoxicillin was used as a positive control (40 μg/ml) for bacteria while Fluconazole (120 μg/ml) was used for fungi. The diluted cultures were spread on sterile Muller–Hinton agar plates. The plates were then incubated at 37°C for 18 to 24 h for bacterial pathogens and 3 days for fungal pathogens. The antimicrobial activity was evaluated by measuring the diameter of inhibition zone. The experiment was carried out in triplicate and the mean of the diameter of the inhibition zones was calculated. Antimicrobial inhibition activities measured were compared to that of Amoxicillin and Fluconazole standards for antifungal (Vlietinck, 1997).
RESULTS AND DISCUSSION
Aerial shoots and roots of C. capensis were used in this study. Extractions and isolations were specifically targeted for alkaloids and flavonoids indicated by the preliminary phytochemical screening results (Babajide et al., 2010) where high concentrations of flavonoid and alkaloids in the methanolic extracts of both the root and the aerial shoot were noted. A total of 6 compounds, viz- 1 to 6 were isolated from the fractionation of the TTA, methanolic extract of the aerial shoot and root. The TLC investigations of these 6 compounds revealed 1, 2 and 3 to be alkaloids as demonstrated by their positive test to dragendorff reagent using berberine as marker, while 4, 5 and 6 were flavonoids due to their positive test to vanillin in sulphuric acid using quercetin as a marker.
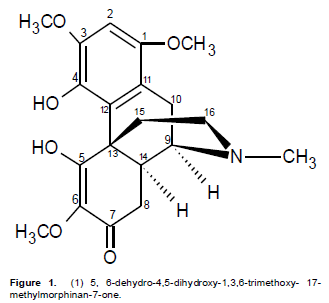
Compound 1 was obtained as a reddish-yellow powder with melting point of 85 to 87°C. The UV spectrum had a lmax values at 222 nm known to be associated with α,β-unsaturated carbonyl chromophore (Stuart, K.L., 1971) and that at 260 nm is typical for an aromatic ring system. The shoulder observed at 304 nm is a general characteristic of 8,14-dihydromorphinandienone alkaloids (Freitas et al., 1995). The IR spectrum showed pronounced bands at 3485 cm-1 (hydroxyl group), 2940 cm-1 (C-H of an olefinic group), 1685 cm-1 (α, β-unsaturated carbonyl), 1615 cm-1 (C=C olefinic) and 1500 cm-1 (C=C aromatic). The El mass spectrum indicated the molecular formula to be C20H25NO6 from the peak observed at M+ (375). The following fragments were observed: 360 (M- CH3) +, 332 (360 – CO) +, 238 (M – C8H902)+ and 42 (H2C=C=O)+, being characteristic of the morphinandienone alkaloids (Wheeler et al.,1968). The complete analysis of 1H NMR, 13C NMR, HMBC, COSY and NOESY were used in assigning the structure of 1. The 1H NMR (400 MHz, pyridine- d5) showed only one aromatic signal at δ6.91 which implies that the aromatic ring is penta-substituted. The four singlets integrated for three protons each were observed at δ 2.26 for (N-Me), with the other three at δ3.69 for MeO-6, 3.76 for MeO-3 and 3.83 for MeO-1. The two single proton signals at 10.75 and 11.50 were attributable to HO-4 and HO-5 respectively.
The 13CNMR (100MHz, pyridine-d5) similarly showed a signal at δ194.7 (s, C-7) for the α,β-unsaturated carbonyl, δ43.1 (s, N-Me), δ55.3 (s, C-6-OMe), δ56.5 (s, C-3-OMe) and δ57.2 (s, C-1-OMe). The complete assignments of all the protons and carbons are given in Table 1. This data was found to be in agreement with the literature (Blasko and Cordell, 1988; Vecchietti et al., 1981) thus confirming compound 1 to be 5,6-dehydro-4,5-dihydroxy-1,3,6-trimethoxy- 17-methylmorphinan-7-one (Figure 1). Compound 1 to the best of our knowledge, has not been reported previously.
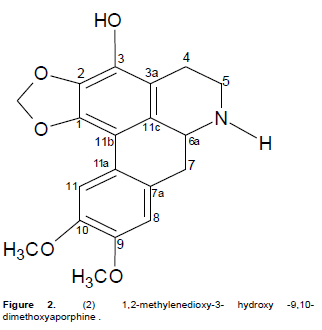
Compound 2 was obtained as a reddish-brown fluffy powder with m.p of 221 to 223°C. Its UV spectrum showed absorptions at 225, 243, 277 and 300 (shoulder) nm which are characteristic of 1, 2, 3-trisubstituted aporphinoid systems (Guinaudeau et al., 1979; Guinaudeau and Bruneton, 1993). The IR spectrum showed bands at 3420, 3050, 2925, 1636, 1458, 1387 and 1060 cm-1 confirming the aporphinic skeleton of 2. The EIMS showed an [M]+ at 341.0112 which suggested the molecular formula of C19H19O5N. 1HNMR, 13CNMR, HMBC, COSY and NOESY spectra were used in assigning the structure. The 1H NMR (400 MHz, C5D5N) spectrum showed an AB set of doublets at δ5.94 and 6.13 (J = 1.2 Hz), characteristic of a methylenedioxy group at positions C-1 and C-2 in aporphines (Guinaudeau and Bruneton, 1993). The presence of an ABCD tetra substituted ring system with signals at δ 7.31 (br s) and 8.32 (br s) is a strong indication that ring D (the dimethoxysubstituted ring) is disubstituted. The 13C NMR (100 MHz, C5D5N) spectrum showed a signal at δ101.3 (s, C-1, 2 methylenedioxy). In the 1H - 1H NOESY spectrum one of the methylenedioxy protons (δ 6.13) showed a correlation with H-11 (δ8.32) while H-8 (δ7.31) correlated with the H-7 methylene protons at (δ2.95-3.10). Complete assignments of all protons and carbon atoms are listed in Table 2. The spectroscopic analysis above established the structure of 2 as 1, 2-methylenedioxy-3- hydroxy -9, 10-dimethoxyaporphine (Figure 2) and to the best of our knowledge it is being reported for the first time.
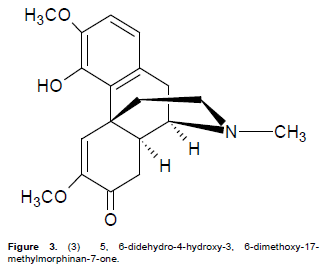
Compound 3 was isolated as a golden brown powder with m.p of 77 to 80°C and runs as a single spot in Hex: CHCl3: MeOH (3:10:2). This spot showed a characteristic orange colour of an alkaloid that is closely similar to the berberine standard TLC plate marker used when sprayed with dragendorff reagent. The IR spectrum showed a pronounced band at 3517 cm-1 due to the presence of a hydroxyl group while that at 2952 cm-1 was assigned to C-H of an olefinic system. The band at 1685 cm-1 is a characteristic of an α,β-unsaturated carbonyl system as observed for 1, the band at1616 cm-1 is due to the olefinic C=C while that at 1510 cm-1 is due to the aromatic C=C bond. Its UV spectrum showed absorptions at 226 nm which is known to be characteristic of an α,β-unsaturated carbonyl chromophore and at 261 nm due to an aromatic ring while the band at 302 nm is a charac-teristic of the 8,14-dihydromorphinandienone alkaloids as was noticed for 1 (Stuart, K.L., 1971). The El mass spectral peak of 329.2133 suggested a molecular formula of C19H23O4N for 3, for which the following fragments were observed: - (1) 314 [M- CH3]+, (2) 286 [314 – CO]+, (3) 192 [M – C8H9O2]+ and (4) 42 [H2C=C=O]+ confirming the characteristics of a morphinandienone alkaloid as observed for 1 (Wheeler et al., 1967). The 1H NMR spectrum showed an AB pair of doublets at δ 6.90 and 6.78 (J = 8.4 Hz) for H-2 and H-1 respectively, while the three 3-proton singlets at δ 2.28 (N-Me), δ 3.62 (MeO-6) and δ 3.74 (MeO-3) (with similar numbering system as shown in 1) were assigned as indicated. The singlet at δ 10.72 was attributable to HO-4. The 13C NMR spectrum showed 19 carbon signals which supported the calculated molecular formula of C19H23O4N. The carbons were assigned as shown in Table 3 where δ 43.3 is for N-Me, δ 55.2 for C6-OMe, δ 56.6 for C3-OMe and δ 194.5 for the C7-C=O. Complete assignments of all the protons and carbons are given in Table 3.
The structure of compound 3 was therefore established to be 5, 6-didehydro-4-hydroxy-3, 6-dimethoxy-17-methylmorphinan-7-one as shown in Figure 3. Literature reveals that 3 has been previously isolated from Croton (Vecchietti et al., 1981) and some Cissampelos species such as C. sympodialis (Freitas et al., 1995) and C. pareira (Amresh et al., 2007). This is however the first time that the alkaloid has been reported as found in C. capensis.
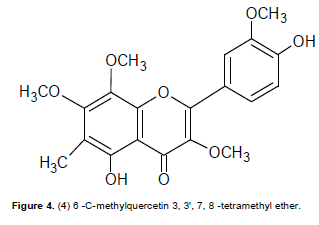
Compound 4 was a yellow amorphous powder with a m.p of 234 – 236oC. It showed EI mass spectral peak (M)+. at m/z 388.0103 suggesting a molecular formula of C20H2008. The IR spectrum showed pronounced bands at 3401 cm-1 (hydroxyl group), 2932 cm-1 (C-H olefinic), 1653 cm-1 (carbonyl group), 1612 cm-1 (C=C olefinic) and 1556 cm-1 (C=C aromatic). The UV spectrum showed two peak maxima at 357 and 255 nm respectively. The band at 271 nm showed a large bathochromic shift in the presence of sodium acetate, and an additional band appeared at 343 nm in the methanol NaOH spectrum which suggested a flavonol with a 7-OMe group. The 1H NMR spectrum showed an aryl methyl singlet at δ 2.14 and four distinct methoxyl singlet peaks at δ 3.74 ( OMe-3), 3.96 (OMe-3'), 4.01 (OMe-7) and 4.56 (OMe-8). The spectrum further showed the presence of only three 1-proton aromatic signals at δ 6.91 (d, J = 2.3 Hz), 7.60 (dd, J = 8.5 and 2.3 Hz) and 7.70 (d, J = 8.5 Hz) suggesting a 3',4'-disubstituted aromatic ring C, and a fully substituted aromatic ring A. The characteristically chelated 5-OH group was observed at δ 12.92 while that of 4’-OH was observed δ 10.90. By careful comparison of the 13C NMR data of 4 with existing literature (Roitman and James, 1985; Breitmeier and Voelter, 1990; Babajide et al., 2008), the 4'-substituent was assigned to a hydroxyl hydroxyl group while that at the 3' position was assigned as a methoxyl group. Corroboration of this deduction was evident from chemical shifts of the substituted carbons, viz., C-3' at δ 147.2 and C-4' at 148.5. The other methoxyl groups were attached at C-3 (δ 137.5), C-7 (δ 145.4) and to C-8 (δ at 149.5 ppm which was in agreement with the HMBC correlations OMe-3/C-3 and OMe-7/C-7 and OMe-8/C-8 (Table 4).
The only non-oxygenated methyl group present was assigned as being attached to C-6, (which appeared at 106.6 ppm in the 13C spectrum). Other peaks as demonstrated by the HMBC cross peaks were OH-5/C-5, C-6, C-4a; Me-6/C-5, C-6, C-7. Other carbon peaks were similarly assigned as shown in Table 4. Based on the above information the structure of 4 was suggested to be 3,7,8,3'-tetramethoxy-6-C-methyl-5,4'-dihydroxyflavone (6-C-methylquercetin 3, 3',7, 8 -tetramethyl ether) as shown in Figure 4 and to the best of our knowledge it is being reported for the first time.
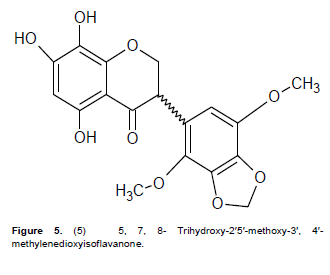
Compound 5 was obtained as a yellow waxy powder with m.p of 185 to 187°C. A molecular formula of C18H18O9 was suggested based on the molecular ion peak obtained at m/z 378.0625 and the fragmentation pattern in the EIMS. An exhaustive comparative study of the data obtained, revealed the general features of an isoflavanone nucleus (Mabry et al., 1970; Markham and Chari, 1982; Rahman and Gray, 2005; Rahman et al., 2007). The UV absorption spectrum with maxima at 333 and 288 nm suggested an isoflavonoid. The IR spectrum had a strong absorbance at 3421 cm−1 due to an O-H group present, 2914 cm−1 for the C=C-H group, and 1634 cm−1 for the C=O system. The 1H NMR spectrum showed an ABC pattern of 1-proton signals viz., δ 4.69 (t, J = 11.0 Hz), δ 4.50 (dd, J = 11, 5.6 Hz) and δ 4.43 (dd, J = 11.0, 5.6 Hz) assigned to the H-2 and H-3 protons respectively, which was similar to that observed in Markham and Chari’s work as reported in 1982 for an isoflavanone system. The 1H NMR spectral data (Table 5) also exhibited three H-bonded groups viz., C-5 hydroxyl at δ 12.89, C-7 hydoxyl at 12.98 and C-8 hydroxyl at δ 13.02. Two 1-proton singlets at δ 6.52 and δ 6.80 were assigned to H-6 of ring A and H-6¢ of ring C of the isoflavanone. The presence of 2 methoxyl groups as well as methylenedioxy group were apparent from their signals at δ 3.96 (2′-OMe), 3.87 (5′-OMe) and δ 5.90 (–OCH2O–) respectively. The 13C NMR spectrum demonstrated that compound 5 contained a total of 18 carbons including a carbonyl group as indicated by the IR absorption peak at 1634 cm−1. The assignment of all carbons and the placement of the 2-methoxyl and methylenedioxy groups within the molecule were achieved by 2D NMR spectroscopic analysis. The HMBC data (Table 6) showed correlation of the C-5 hydroxyl group with a methine at δ 97.7 and a quaternary carbon at δ 103.3 which were assigned to C-6 and C-4a respectively.
The signals at δ 164.7, 168.6 and 169.3 were assigned to C-8a, C-7, and C-8 respectively as shown in Table 5. Correlation of the methoxyl protons with the carbons at δ 142.5 and 145.6 in the HMBC data proved the presence of C-2′ and C-5′ methoxy groups at these positions. The methylenedioxy group exhibited J3 correlation with carbons at δ 137.8 and 149.8 and the occurrence of a J3 correlation of H-6′ to δ 149.8 suggested location of the methylenedioxy between C-3′ and C-4′. The assignment of C-6′ at δ 125.1 was also confirmed by the coupling observed in the HMQC which suggested the methine carbon at δ 48.5 as C-3 as observed in the J3 for H-6′. Furthermore, the peak at δ 71.1 was assigned to C-2 as it showed a J2 correlation to H-3 in the HMBC spectrum, and direct coupling with H-2 in the HMQC spectrum. The COSY spectrum showed correlation between H-3 and the two non-equivalent protons at C-2. The large diaxial coupling (J = 11.0 Hz) observed between H-3 and H-2 placed the aryl substituent at C-3 in the pseudo-equatorial position. Based on the above information the structure of compound 5 was therefore suggested to be 5,7,8-trihydroxy-2′,5′-dimethoxy-3′,4′methylenedioxy-isoflavanone (Figure 5) and to the best of our knowledge it is being reported for the first time.
Compound 6 was isolated as a creamy yellow powder was assigned as C17H1408 based on the molecular ion peak at m/z 346.0752. The UV spectrum with maxima at 351 and 260 nm suggested a flavonol with a 7-OH group. A deep broad band observed in the IR spectrum at 3500 to 3400 cm-1 was indicative of the presence of a hydrogen-bonded hydroxyl groups. The 1H NMR spec-trum exhibited two 3-proton singlets assignable to one aryl methyl group at δ 2.11 (Me-6), very similar to that of 4 and one methoxyl group at δ 3.87 due to 3-OMe. The aromatic region showed three 1-proton signals typical of a 3', 4'-disubstituted ring C similar to that observed in 4 viz., δ 7.03 (d, J = 2.3 Hz), δ 7.62 (dd, J = 8.6 and 2.3 Hz) and δ 7.75 (d, J = 2.3 Hz). A broad cluster of 1-proton singlets at δ 10.23 (4'-OH), 11.15 (3'-OH), 12.05 (8-OH), 12.90 (7-OH) and 13.05 (5-OH) were due to five OH groups. The 13C spectrum (Table 7) demonstrated a pattern similar to that of compound 4, in which the C ring is di-substituted, carrying two hydroxyl groups at C-3' (163.6) and C-4'(164.5). The HMBC data for ring A was expected to be slightly different from that of 4 with the C-7(159.2) position showing the presence of an OH group. The HMBC cross peaks observed for OH-5/C-5, C-6, C-4a and Me-6/C-5, C-6, C-7, OH-8/C-7, C-8, C-8a indicated that the OH groups were located at C-7 and C-8 due to their respective coupling effects. The location of the single available methoxyl group and methyl group at C-3 and C-6 respectively, were further verified by their HMBC correlations with C-3 and C-6 respectively. Consequently the structure of 6 was with m.p of 217 to 219°C and the molecular formula of 6 assigned as 3-methoxy-6-C-methyl-3',4',5,7,8 -pentahydroxyflavone (6 -C- methylquercetin -3-methyl ether) (Figure 6). Compound 6 has, to the best of our knowledge, not been reported previously. It should be noted that there were several other extracts, fractions and subfractions that were not further analyzed due to insufficient quantities and time.
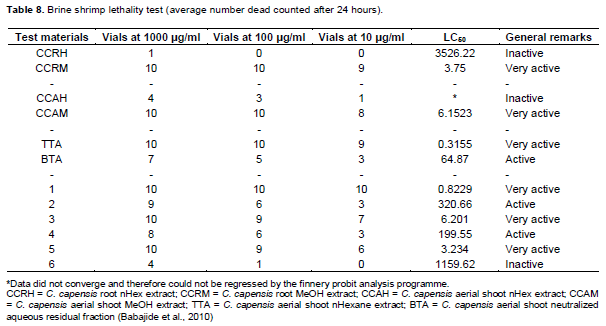
Brine shrimp lethality bioassay
The brine shrimp lethality bioassay as described earlier was performed on all the extracts and fractions as shown in Babajide et al. (2010) and also on the isolates (1 to 6) estimating their LC50 with 95% confidence. As shown in Table 8, where three test bands were used viz., Inactive (LC50 > 700), active (LC50 < 700) and very active (LC50 < 10). The results demonstrated that all the hexane extracts showed no activity. Clearly evident was the finding that the highest bioactivity appeared in the methanolic extracts, both in the root and the aerial shoot of this plant. The TTA showed a very high active value of 0.3155 (Table 8) thus indicating better activity than the BTA with a value of 64.87. This could be correlated with the isolation of the highly active compound 1 from the TTA. All the extracts and isolates (1, 3 and 5) that were very active should be considered as good candidates for antimicrobial and antiviral agents, since the lethality of a test substance to brine shrimp nauplii has been linked to the possible ability of such substances to kill cancer cells (antitumor activity) and possession of anti-inflammatory properties (Mc Laughlin et al., 1991; Farthing, 2000). However, this does not mean that the inactive com-pounds (6 and hexane extracts) would not be useful for some other purposes such as a good antioxidant agents.
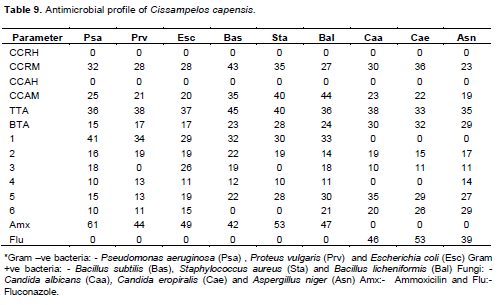
Antimicrobial activity evaluations
The antimicrobial evaluation was carried out using the diffusion method in which three organisms were used in each case. For gram-negative bacteria: P. aeruginosa, P. vulgaris and E. coli Sero type 1 were used while for gram-positive, B. subtilis, S. aureus and B. licheniformis were used. The fungal species used were C. albicans, C. eropiralis and A. niger. The assay was set up as des-cribed earlier. The inhibition zones for both the bacteria and the fungi were measured in triplicate for all the extracts and the 6 identified isolates. The average was determined and compared to the positive controls used (Amoxicillin; 40μg/ml for bacteria and Fluconazole; 120μg/ml for fungi). Similar patterns were observed in the microbial analysis except in very few cases as shown in Table 9. The selection of the three (3) sets of test organisms was designed to allow for the study of a reasonably wide spectrum of antimicrobial activities of the extracts and isolates.
The results showed different levels of activity for both the extracts and the isolates. Any value > 20 mm is assumed to be moderately active. There was more antibacterial than antifungal activity. The highest activity was recorded for the gram +ve bacteria while that of gram –ve bacteria was more than the fungi. The maxi-mum zone (45 mm) of antibacterial activity was observed in the TTA fraction against the gram +ve organism B. subtilis. This may be due to the strong antimicrobial and antiviral activities of the alkaloids in the plant that is well known for the treatment of malaria, fungal infections, inflammations and cancer (Kaur et al., 2009; Mc Gaw et al., 2000). No activity was recorded for the hexane extracts against all nine organisms. High values of acti-vity were recorded for the drug resistant breed of bacteria P. aeuruginosa, where an inhibition zone of 36 mm was recorded for TTA, 25 mm for CCAM and 32 mm for CCRM.
Some preferential selective activity was observed in which the compound preferentially act on certain selected organism were inactive against the other set (Babajide et al., 2010; Shai et al., 2008). Similar observations were made in the isolated compounds, where high values were recorded for 1 against gram +ve and –ve bacteria whereas there was no effect on all the three fungi. High fungicidal effect was recorded for 5 while the activities were generally low for compound 4. The observed biological activities of extracts from this plant may thus be due to the presence of both alkaloids and flavonoids, as these compounds are generally known to exhibit a wide range of activities which include anti inflammatory, antithrombotic, anticancer and antiviral, where some of these may be associated with their ability to scavenge free-radicals (Babajide et al., 2008; Shai et al., 2008; Rahman et al., 2007; Liu et al., 1990).
CONCLUSION
This study successfully relates the specific constituents present in C. capensis to its widespread ethnomedicinal uses. The results provide a useful profile of active compounds for the development of useful and promising anticancer, antimicrobial and antioxidant agents in the future.
ACKNOWLEDGEMENTS
The authors appreciate the National Research Foundation (NRF) of South Africa for the research funding, Prof Bouic P.J.D. and Mrs Oduwole E.O. of Synax both of Department of Medical Microbiology, Faculty of Health Sciences, Stellenboch University, South Africa for providing the microbial pathogens used, Mr N Coldery of the Medical biosciences department, University of the Western Cape for assisting in some of the microbial assays and Mr Franc Weitz of the Department of Biodiversity and Conservation Biology of the University of the Western Cape for the identification, authentication and collection of the plant materials.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
| Akerele O (1988). Medicinal plants and primary health care: an agenda for action. Fitoterapia 59(5):355-363. | ||||
|
Amresh G, Rao CV, Singh PN (2007a). Antioxidant activity of Cissampelos pareira on benzo(a)pyrene-induced mucosal injury in mice. Nutri. Res. 27:625-632. Crossref |
||||
|
Amresh G, Reddy GD, Rao CV, Singh PN (2007b). Evaluation of anti-inflammatory activity of Cissampelos pareira root in rats. J. Ethnopharmacol. 110:526-531. Crossref |
||||
|
Amresh G, Reddya GD, Rao CV, Shirwaikar A (2004). Ethnomedical value of Cissampelos pareira extract in experimentally induced diarrhea. Acta Pharm. 54(1):27-35. Pubmed |
||||
|
Amresh G, Singh PN, Rao CV (2007c). Antinociceptive and antiarthritic activity of Cissampelos pareira roots. J. Ethnopharmacol. 111:531-536. Crossref |
||||
|
Babajide JO, Babajide OO, Daramola AO, Mabusela WT (2008). Flavonols and an oxychromonol from Piliostigma reticulatum. Phytochemistry 69:2245-2250. Crossref |
||||
| Babajide JO, Mabusela WT, Green IR, Ameer F, Weitz F, Iwuoha EI (2010). Phytochemical screening and biological activity studies of five South African indigenous medicinal plants. J. Med. Plants Res. 4(18):1924-1932. | ||||
|
Barbosa-Filho JM, Ca-Cunha EVL, Gray AI (2000). Alkaloids of the Menispermaceae. In: Cordell GA (Ed.), The Alkaloids, v. 54. Academic Press, Illinois, USA. pp. 1-190. Crossref |
||||
|
Barbosa-Filho JM, Da-Cunha EVL, Cornelio ML, Silva Dias CD, Gray AI (1997). Cissaglaberrimine, an aporphine alkaloid from Cissampelos glaberrima. Phytochemistry 44:959-961. Crossref |
||||
|
Blasko G, Cordell GA (1988). Morphinandienone Alkaloids. Heterocycles 27(5):1269-1300. Crossref |
||||
| Botha DJ (1980). The identity of Antizoma harveyana Miers ex Harv. and A. capensis L.f. Diels. J. South Afr. Bot. 46(1):1-5. | ||||
| Breitmeier E, Voelter W (1990). Carbon-13 NMR Spectroscopy, VCH, New York. P 451. | ||||
| Bruneton J (1995). Pharmacognosy and Phytochemistry of Medicinal Plants, second ed. Intercept Ltd., Hampshire. pp. 385-386. | ||||
| Christophersen C, Larsen C, Dimayuga RE (1991). Traditional Medicine-A potential resource exploitation of natural products. The H.C. Orsted institute, Copenhagen. pp. 8-12. | ||||
| Cillie AM (1992). Kruie op Witblits, Rate, Respte en Feite. Unpublished notes, Worcester Museum. | ||||
| De Wet H (2006). An ethnobotanical and chemotaxonomic study of South African Menispermaceae. PhD Thesis, University of Johannesburg. | ||||
|
De Wet H, Van Heerden FR, Van Wyk BE (2004). Alkaloids of Antizoma angustifolia (Menispermaceae). Biochem. Syst. Ecol. 32:1145-1152. Crossref |
||||
|
De Wet H, Van Heerden FR, Van Wyk BE (2005). Alkaloids of Antizoma miersiana (Menispermaceae). Biochem. Syst. Ecol. 33:799-807. Crossref |
||||
|
De Wet H, Van Wyk BE (2008). An ethnobotanical survey of southern African Menispermaceae. South Afr. J. Bot. 74:2-9. Crossref |
||||
| Dictionary of Natural Products (1996). Release 4:2. Chapman and Hall, London (CD289 ROM). | ||||
| Dictionary of Natural Products on CD-ROM (2006). Chapman Hall, CRC Press, Hampden Data Services, Ltd. | ||||
| Dykman EJ (1891). Kook-Koek- en Resepte Boek. Paarl: Paarlse Drukpers Maatskappy. | ||||
|
Farthing MJ (2000). Diarrhoea: a significant worldwide problem. Inter. J. Antimicrob. Agent 14:65-69. Crossref |
||||
|
Freitas MR, Alencar JL, Da-Cunha EVL, Barbosa-filho JM, Gray AI (1995). Milonine, an 8,14-dihydromorphinandienone alkaloid from leaves of Cissampelos sympodialis. Phytochemistry 40(5):1553-1555. Crossref |
||||
|
Ganguly M, Borthakur MK, Devi N, Mahanta R (2007). Antifertility activity of the methanolic leaf extract of Cissampelos pareira in female mice. J. Ethnopharm. 111:688-691. Crossref |
||||
|
Gessler MC, Msuya DE, Nkuyuna MHH, Mwasunmbi LB, Schar A, Heinrich M, Tanner M (1995). Traditional healers in Tanzania: The treatment of malaria with plant remedies. J. Ethnopharm. 48(3):131-144. Crossref |
||||
|
Graham JG, Quinn ML, Fabricant DS, Farnsworth NR (2000). Plants used against cancer – an extension of the work of Jonathan Hartwell. J. Ethnopharm. 73:347-377. Crossref |
||||
| Guinaudeau H, Bruneton J (1993). Isoquinoline alkaloids. Alkaloids and Sulphur Compounds. Methods in Plant Biochemistry 8 Waterman, P. G., ed., London Academy Press. pp. 373-419. | ||||
|
Guinaudeau H, Lebouef M, Cavé A (1979). Aporphine Alkaloids II. J. Nat. Prod. 42(4):325-360. Crossref |
||||
|
Hamill FA, Apio S, Mubiru NK, Bukenya-Ziraba R, Mosango RM, Maganyi OW, Soejarto DD (2003). Traditional herbal drugs of Southern Uganda, II: literature analysis and antimicrobial assays. J. Ethnopharm. 84:57-78. Crossref |
||||
| Iwu MM (1993). Handbook of African Medicinal Plants. C.R.C. Press, Boca Raton, Florida. pp. 26-267. | ||||
|
Jittra S, Suwayd N, Steve WC, Douglas GH (2005). Extraction and physicochemical characterization of Krueo Ma Noy pectin. Food Hydrocol. 19:793-801. Crossref |
||||
|
Kaur K, Jain M, Kaur T, Jain R (2009). Antimalarials from nature. Bioorg. Med. Chem. 17:3229-3256. Crossref |
||||
|
Liu S, Babajide O, Charles DH, Alvie M (1990). 3-Methoxysampangine, a novel antifungal copyrine alkaloids fungi deistopholis pattern. Antimicrob. Agent Chemother. 34(4):529-533. Crossref |
||||
|
Mabry TJ, Markham KR, Thomas MB (1970). The Systematic Identification of Flavonoids. Springer Verlag., New York. P 2204. Crossref |
||||
|
Mander M (1998). Marketing of indigenous medicinal plants in south Africa. In: A Case study in Kwa-Zulu Natal. FAO, United Nations, Rome. Pubmed |
||||
|
Markham KR, Chari VM (1982). Carbon-13 NMR spectrosocopy of flavonoids. In: Harborne JB, Mabry TJ (Eds.), The Flavonoids: Advances in Research. Chapman and Hall, London, UK. pp. 19-134. Crossref |
||||
|
McGaw LJ, Jager AK, van Staden J (2000). Antibacterial, anthelmintic and anti-amoebic activity in South African medicinal plants. J. Ethnopharm. 72:247-263. Crossref |
||||
| Mc Laughlin JL, Chang CJ, Smith DL (1991). Bench – top bioassay for the discovery of bioactive natural products. An update. Attamra (Ed), studies in natural product chemistry. Elservier Science Publishers B.V, Amsterdam. pp. 383-409. | ||||
|
Pillay P, Maharaj VJ, Smith PJ (2008). Investigating South African plants as a source of new antimalarial drugs. J. Ethnopharm. 119:438-454. Crossref |
||||
|
Rahman MM, Gibbons S, Gray AI (2007). Isoflavanones from Uraria picta and their antimicrobial activity. Phytochemistry 68:1692-1697. Crossref |
||||
|
Rahman MM, Gray AI (2005). A benzoisofuranone derivative and carbazole alkaloids from Murraya koenigii and their antimicrobial activity. Phytochemistry 66:1601-1606. Crossref |
||||
|
Ramirez I (2003). Cissampeloflavone, a chalcone-flavone dimer from Cissampelos pareira. Phytochemistry 64(2):645-647. Crossref |
||||
| Rasoanaivo P, Ratsimamanga-Urverg S (1993). Biological Evaluation of Plants with Reference to the Malagasy Flora. Napreca, Madagascar. pp. 9-83. | ||||
|
Roitman JN, James LF (1985). Chemistry of toxic range plants. Highly oxygenated flavonol methyl ethers from Gutierrezia microcephala. Phytochemistry 24(4):835-848. Crossref |
||||
| Rood B (1994). Uit die veldapteek. Tafelberg, Cape Town. | ||||
|
Sanchez-Medina A, García-Sosa K, May-Pat F, Pe-a-Rodríguez LM (2001). Evaluation of biological activity of crude extracts from plants used in Yucatecan Traditional Medicine Part I. Antioxidant, antimicrobial and β-glucosidase inhibition activities. Phytomedicine 8(2):144-151. Crossref |
||||
|
Shai LJ, McGaw LJ, Aderogba MA, Mdee LK, Eloff JN (2008). Triterpenoids with antifungal and antibacterial activity from Curtisia dentata (Burm.f) C.A. Sm. Leaves. J. Ethnopharmacol. 119:238-241. Crossref |
||||
|
Sloan A, Deborah LZ, Kenneth M, Joanne SP, Christine MB, Terry M, Robert B, Seef P, Dennis S, Donald T, Sheo BS (2007). Anthelmintic Activity of Aporphine Alkaloids from Cissampelos capensis. Planta Med. Lett. 73(3):296-297. Crossref |
||||
| Smith CA (1966). Common Names of South African Plants. Memoirs of the botanical survey of South Africa, Vol. 35. Department of Agriculture & Technological Service. The Government Printer, Pretoria. | ||||
|
Ssegawa P, Kasenene JM (2007). Medicinal plant diversity and uses in the Sango bay area, Southern Uganda. J. Ethnopharm. 113:521-540. Crossref |
||||
|
Stuart K L (1971). Morphinandienone alkaloids. Chem. Rev. 71:47-72. Crossref |
||||
| Taylor L (2005). The Healing Power of Rainforest Herbs, Square One Publishers Inc., USA. | ||||
|
Van Wyk BE (2008). A review of Khoi-San and Cape Dutch medical ethnobotany. J. Ethnopharm. 119:331-341. Crossref |
||||
|
Van Wyk BE, De Wet H, Van Heerden FR, (2008). An ethnobotanical survey of medicinal plants in the south-eastern Karoo, South Africa. South Afr. J. Bot. 74:696-704. Crossref |
||||
| Van Wyk BE, Gericke N (2000). People's Plants: A Guide to Useful Plants of Southern Africa. Briza Publications, 1st ed. Pretoria, South Africa, ISBN: 978 1 875093 19 9. | ||||
| Van Wyk BE, Van Oudtshoorn B, Gericke N (1997). Medicinal Plants of South Africa 2nd improved impression, 2000. Briza Publications, Pretoria. | ||||
|
Vecchietti V, Casagrande C, Ferrari G, Danieli B, Palmisano G (1981). Alkaloids of Ocotea acutangula. J. Chem. Soc. Perkin Transact. 1:578-581. Crossref |
||||
| Vlietinck AJ (1997). Biologically active substances from traditional drugs. In: Hostettman K, Lea PJ (Eds.), Proceeding of the Phytochemical Society of Europe on Biologically Active Natural Products. Oxford University Press, Oxford. pp. 33-47. | ||||
|
Wagner H, Bladt S (2001). Plant Drug Analysis: a thin layer chromatography Atlas, second edition. Springer, New York. pp. 349-364. PMid:11439368 |
||||
| Watt JM, Breyer-Brandwijk MG (1962). Medicinal and Poisonous Plants of Southern and Eastern Africa, 2nd ed. Churchill Livingstone, Edinburgh – London. pp. 53-54, 325, 450-451. | ||||
|
Wheeler DMS, Kinstle TH, Rinehart KL (1968). Additions and corrections-mass spectral studies alkaloids related to morphine. J. Am. Chem. Soc. 90(21):5947-5947. Crossref |
||||
| World Health Organisation (WHO) (2002). WHO Traditional Medicine Strategy 2002–2005.World Health Organisation, Geneva, WHO/EDM/TRM/2002.1. | ||||
| World Health Organisation (WHO) (2005). World Malaria Report http://www.rbm.who.int/wmr2005/pdf/adve.pdf. | ||||
|
Wu SJ (2007). Tetrandrine inhibits proinflammatory cytokines, iNOS and COX-2 expression in human monocytic cells. Biol. Pharm. Bull. 30(1):59-62. Crossref |
||||
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


