Full Length Research Paper

ABSTRACT
The purpose of this study was to apply an intentional mismatch design technique for primer-based detection of gene targets. In this study, wild-type versus mutant cystic fibrosis transmembrane conductance regulator alleles as well as two related adenovirus B7 and E4 genomes were used to test discrimination of primers. Lambda viral DNA genome template as well as published primers versus purified human genome template was used as controls. The presence of the ΔF508 mutation and adenovirus B7 could be accurately detected using allele-specific primers, with the reverse ΔF508 primers containing an intentional mismatch, to increase the steric hindrance, therefore both diagnosing CFTR and associated viruses.
Key words: cystic fibrosis, genetic diagnosis, assay, PCR, electrophoresis.
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive genetic disorder that can be caused by over 900 different mutations in the CFTR gene (Roqué et al., 2001). Patients with cystic fibrosis experience a range of symptoms, most common of which is thick, viscous mucus in the lungs, a result of defective chloride channels. The consequential buildup of mucus in the lungs provides a location for many types of bacteria as well as potential viruses, such as the human adenovirus, which could increase the severity of infections for affected patients (Miró-Cañís et al., 2017).
The focus of this assay is the ΔF508 mutation. The mutation is a 3 base-pair deletion within the CFTR gene. This deletion impacts two codons but creates an alternate coding for the original amino acid, isoleucine, and thus only the removal of the amino acid phenylalanine occur. This deletion results in the improper folding of the nascent CFTR protein, ultimately preventing the ion channel from reaching the apical membrane (Rafeeq and Murad, 2017).
Prior research suggests that the identification of the ΔF508 mutation is an effective method of diagnosing the majority (63.9%) of patients with CF (Marson et al., 2013). There are many methods for diagnosing CF, such as sweat chloride tests, reverse blot dot hybridization, and polymerase chain reaction (PCR) (Dooki et al., 2011). PCR was the method used in this study, and gel electrophoresis was used to visualize products signifying in order to identify the presence of CF and human adenovirus B7 (Garibyan and Avashia, 2014). Adenovirus B7 is one of the most prominent causes of severe respiratory infections in cystic fibrosis patients and therefore was chosen to be the focus of a second assay
(Metzgar et al., 2007).
This assay allows for a more specific identification of the human adenovirus B7 (Xu and Erdman, 2001). The purpose of this study was to evaluate ways to potentially improve the accuracy and reliability of CF screenings, as well as the detection of a viral infection by engineering primers that bind to specific mutations/strains. Allele specific primers with an intentional mismatch on the 3’ end to detect the ΔF508 mutation in the CFTR gene were developed and tested. The intentional mismatch promoted allele-specific binding through increased steric hindrance, to reduce false positives (Yaku et al., 2008). When used in conjunction with a PCR assay, the primers efficiently amplified the appropriate DNA sequences to indicate the presence of the ΔF508 mutation or B7 viral infection.
MATERIALS AND METHODS
Unless otherwise indicated, chemicals were obtained from Sigma (St. Louis, MO). Oligonucleotide DNA primers for polymerase chain reaction were obtained from Integrated DNA Technologies (Coralville, IA). Taq polymerase enzyme was obtained from Syd Labs (Southborough, MA). For molecular weight standard in gel electrophoresis, the 100bp DNA Ladder was obtained from Gold Biotechnology (St. Louis, MO). DNA samples of human Adenovirus B7 (HAdV-B7) and human Adenovirus E4 (HAdV-E4) were obtained from Dr. Adriana E. Kajon at the Lovelace Respiratory Research Institute in Albuquerque, New Mexico.
Primer design
Primers were designed with data from prior literature and use of the Cystic Fibrosis Mutation Database. Primers were created for targeting both mutant and wild-type DNA sequences, including two reverse and one common forward primer (Table 1). For all primers, the G/C content was designed to be approximately 40%, and length 20 base pairs. The ΔF508-CFTR mutation is found at amino acid position 508, located in the N-terminal cytoplasmic NBD1, of chromosome 7 (Thibodeau et al., 2010). Designed mutant-seeking reverse primers (DMR) were engineered to anneal to the CFTR DNA sequence only when the three-base deletion (ΔF508-CFTR) was present. The mutant-seeking primer had an intentional mismatch to reduce non-specific annealing to wild-type DNA. Designed wild-type-seeking primers (DWR) were engineered to anneal to the same locus only when wild-type CFTR sequence was present. The wild-type primer has an intentional mismatch designed to reduce the chance it could anneal to mutant DNA. The primers were also designed to have a G or C base on the final 3’ end position to reduce mis-bonding. A common forward primer was designed to anneal 989 base pairs upstream.
DNA isolation
Samples of wild-type and ΔF508 mutant buccal cells were obtained from human saliva (Mulot et al., 2005). A 10% solution of Chelex-100 resin was used (0.10 grams of chelex resin 1.0mL of DNase free water), to extract genomic DNA. A 1.5mL of sample of approximately 1x106 buccal cells was centrifuged at 14000 rpm for 2 min until a pellet was formed.
The supernatant was removed and a solution of 500µL of 10% chelex was added and vortexed until the pellet was resuspended.
The sample was incubated in a 56ºC water bath for 10 min, and was vortexed for 5 s every 5 min. After 10 min, the sample was placed into a 100ºC sand bath for 5 min. The solution was centrifuged one last time at 14000 rpm (13147 RCF g) for 5 min to reform the pellet. The supernatant with genomic DNA was removed and stored at -20ºC. To quantify concentrations of human DNA samples, a 260/280 analysis was performed with a BioTek Epoch Microplate (nanodrop cassette) UV spectrophotometer.
Polymerase chain reaction
After obtaining the purified DNA, a polymerase chain reaction was utilized to amplify specific target regions of each DNA genome. PCR was conducted with a mixture containing 5µL 10x PCR buffer (containing 200mM Tris-HCl and 500 mM KCl), 10mM dNTPs (deoxynucleotide triphosphates) (1µL), 4.5µg/µL DNA template (1µL), 5U/µL Taq polymerase (1µL), 25nmol forward primer (1µL), 25nmol reverse primer (1µL), 50mM MgCl2 (1µL), and 39µL deionized water. Multiple PCR trials were run amplifying the following: lambda virus with Rz gene-seeking primers, wild-type DNA with published wild-type primers (PWF and PWR) and designed wild-type reverse primers (DWR) in combination with the common forward primers (DCF), mutant DNA with designed mutant reverse primers (DMR) with DCF primers, HAdV-B7 DNA with designed adenovirus B7 primers (DABF and DABR) as positive controls. As for negative controls, wild-type DNA was run with DMR and DCF, mutant DNA with DWR and DCF, and HAdV-E4 DNA with DABR and DABF.
The base protocol for PCR was an initial denaturation at 95? for 3 min, followed by 30 cycles of 95? for 45 s, annealing temperature for 30 s, and 72? for 90 s, and a final extension at 72? for 5 min. For the designed primers, the same procedure was utilized with indicated annealing temperatures, and BioRad T-100 thermocycler was used to facilitate the temperature changes and cycles.
Gel electrophoresis
A 1.5% agarose gel with 5mM lithium 10mM borate buffer was created to visualize all PCR products. The gel consisted of 0.6g agarose, 38mL of dH2O, 2mL of 20x LB buffer, and 4µL of 10,000x SYBR-safeTM dye. The solution was mixed and warmed until dissolved. After some cooling, SYBR-safeTM dye was then added and the solution was cast into a tray mold for polymerization. Gold Bio 100bp ladders were used as reference. Each gel was run at 180 volts for a duration of 20 minutes, or until the loading dye traveled approximately two thirds of the way down the gel, to ensure clear visibility. An ultraviolet trans illuminator system was used to visualize and document resulting DNA bands.
RESULTS
The purpose of this study was to design and evaluate DNA primer sequences created for AS-PCR detection of the ΔF508-CFTR allele as well as for B7 adenovirus (Figure 1 and Table 1).

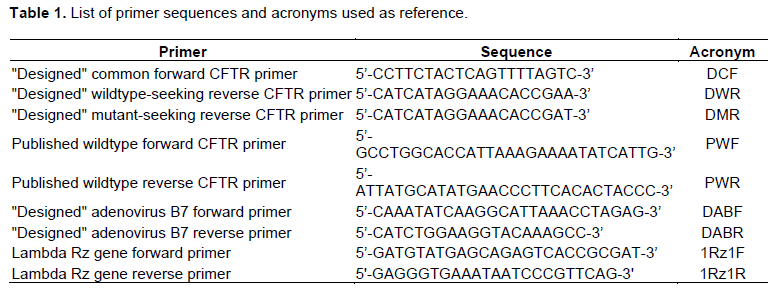
Amplification from Lambda genome served as a routine positive PCR control
A positive PCR control targeted the Rz gene of the Lambda bacteriophage genome. The forward and reverse primers for the lambda DNA were designed to amplify 395 bp segment (Figure 2 Left). Based on the semi-log analysis of the band in lane 4 compared to MW ladder (Figure 2 Right), the calculated band length of ~300+ bp was deemed supportive of successful amplification of lambda, and that PCR reagents and protocols were functioning as nominal.
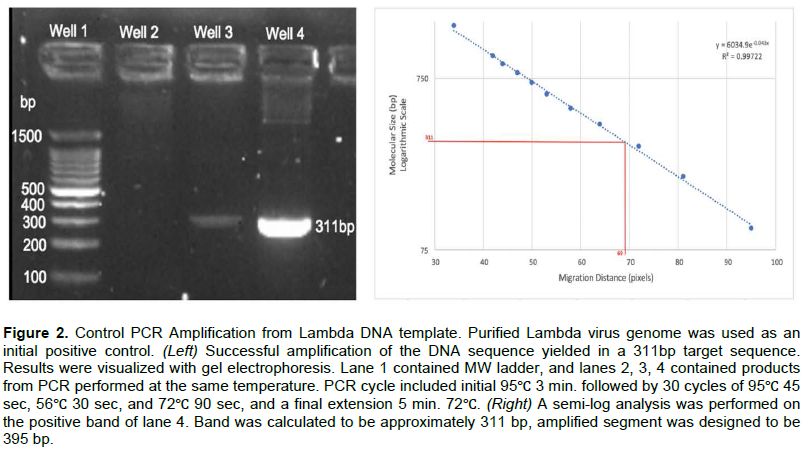
Amplification from human genome served as a control for CFTR gene integrity
Both mutant and wild-type DNA was extracted from buccal cells from human subjects. The buccal cell genomic DNA was purified using Chelex-100 resin. Samples of purified DNA were regularly evaluated for initial genome integrity via visualization in gel electrophoresis, as well as for CFTR gene integrity via controlled PCR amplification.
When high molecular weight genomic DNA was visualized (Figure 3 Left) the initial screen was considered supportive for genome integrity. Published wild-type seeking primer sequences were used to support the presence of intact CFTR gene in genome DNA (Figure 3 Center). A semi-log analysis performed on the amplified band (Figure 3 Right) when calculated to be ~140 bp was considered supportive of successful amplification of human CFTR gene segment in the genomic sample (expected PCR product length was 146 bp).
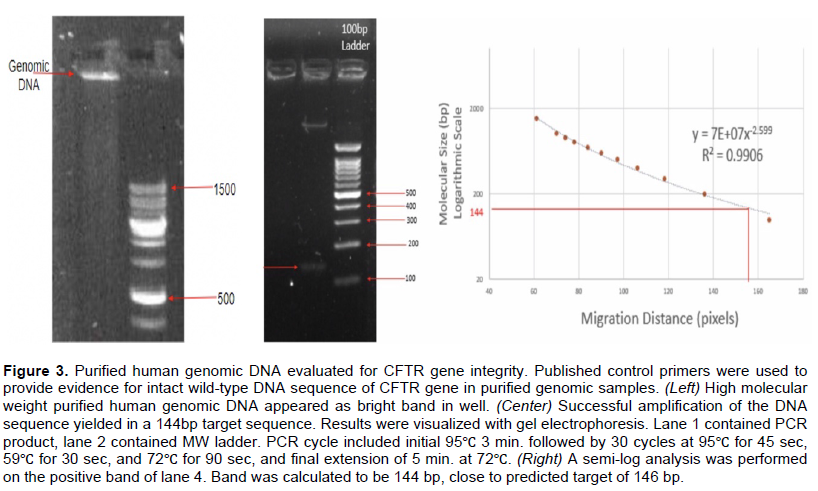
Amplification of wild-type CFTR with wt-seeking primers yielded appropriate product
Designed wild-type reverse primer (DWR) was used in positive and negative contexts. Under conditions that should yield a positive result, wild-type DNA was targeted with DWR and DCF primers (Table 1). The brightest band appeared in lane 4 at an annealing temperature of 47? (Figure 4 Left). A semi-log analysis calculated the band to be ~1000+ bp (Figure 4 Right) indicative of successful amplification of the 989 bp segment as designed. For a negative control, human genomic DNA homozygous for ΔF508-CFTR alleles was amplified with DWR and DCF. No bands appeared.
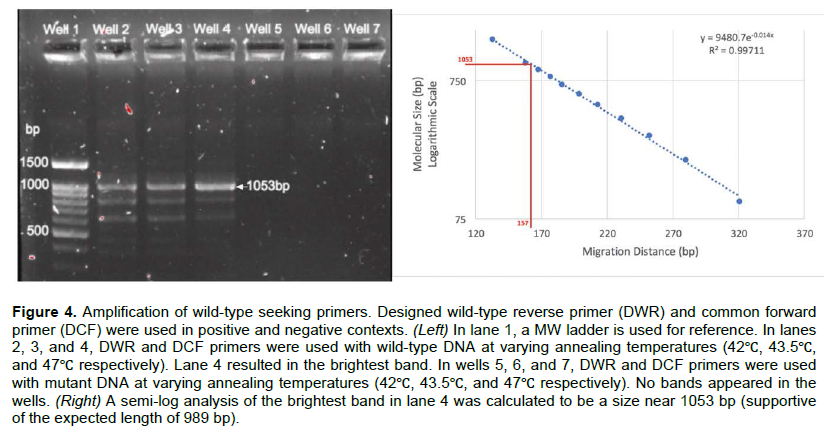
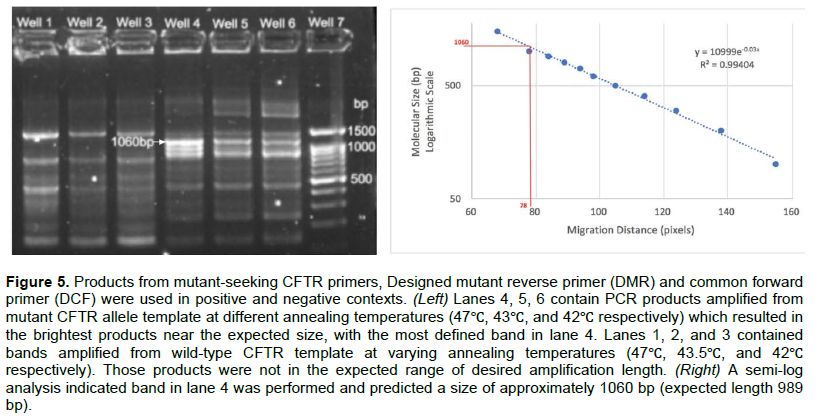
Amplification of mutant ΔF508-CFTR with mt-seeking primers yielded a range of products
Designed mutant reverse and forward primers (DMR/F) were tested in positive and negative diagnostic scenarios. DMR and DMF primers were designed to amplify 989 base pairs versus the ΔF508-CFTR allele. The amplification products using mutant-seeking primers versus mutant ΔF508-CFTR containing template DNA yielded the brightest of bands in lane 4 at an annealing temperature of 47? (Figure 5 Left). The band was calculated to be ~1000+ bp (Figure 5 Right) supportive of a successful annealing, yet among non-specific amplification products. DMR and DMF primers versus wild-type CFTR template DNA yielded only non-specific amplification products and thus DMR and DMF primers require further evaluation.
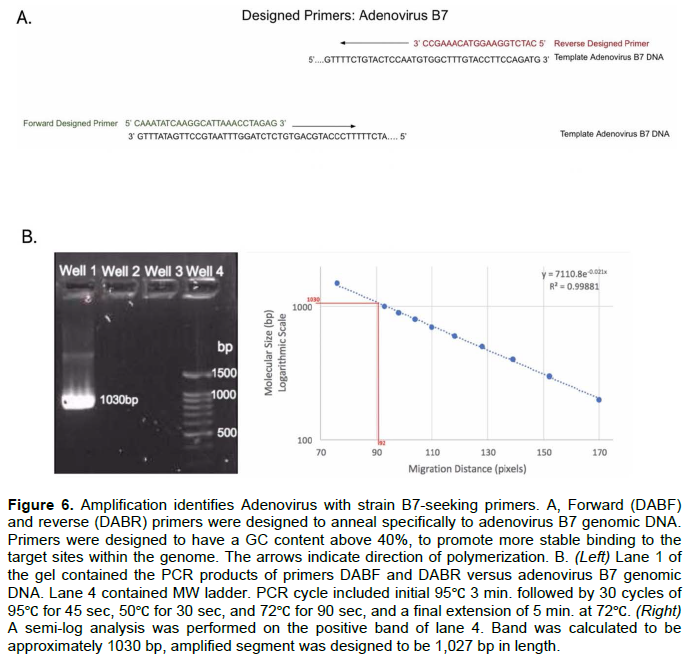
Amplification of adenovirus B7 with B7-seeking primers yielded appropriate products
Primers targeting adenovirus B7 were designed to amplify a 1027 bp segment of the target region (Figure 6A). PCR primers were tested versus adenovirus B7 DNA template for a positive diagnostic, and against adenovirus E4 DNA as a negative one (Figure 6B). The PCR assay resulted in a product calculated at ~1030 base pairs in length for the positive sample and no amplification for the negative sample which suggests that accurate discrimination was obtained.
DISCUSSION
The specific mutation studied, ΔF508-CFTR, is the most common of the cystic fibrosis-related mutations (Marson et al., 2013). The ΔF508 mutation is located on exon 10 of the CFTR gene on chromosome 7 and leads to the deletion of the amino acid, phenylalanine, from the expressed protein (Rafeeq and Murad, 2017). As a result, the protein folding sequence is altered and it is marked for degradation in the endoplasmic reticulum (Thibodeau et al., 2010). Without CFTR reaching the apical membrane, chloride ions, and consequently water, are unable to pass through the apical membrane. The inability of these substances to pass through the body’s epithelial cells leads to the various symptoms experienced by CF patients including, but not limited to: mucus build-up in the lungs, abnormally “salty” sweat, and Distal Intestinal Obstruction Syndrome (DIOS) (Rafeeq and Murad 2017). The surplus of thick mucus in the lungs provides a hospitable environment for many bacteria and for viruses as well, specifically within pediatric population (Miró-Cañís et al., 2017).
The purpose of the authors’ research was to utilize PCR and allele-specific primers in order to test more accurate strategies to identify the presence or absence of the ΔF508 allele in a human sample. As an additional measure to increase specificity, of the ΔF508 reverse primers contained an intentional mismatch on the 3’-end which provided additional steric hindrance between non-complementary DNA to decrease the chances of Taq polymerizing a non-specified strand. In addition to an assay for ΔF508-CFTR, the application of similar techniques was used to pilot a new assay to diagnose the presence of adenovirus B7.
As one regular control, a segment of the Rz gene of lambda viral bacteriophage genomic DNA was amplified via PCR with established primers to provide regular checks that could ensure that reagents and equipment continued to operate optimally. The authors calculated that a 395 base pair segment should be amplified (Taylor et al., 1983) and that result was repeatably supported.
A second control was regularly used to assess and confirm the quality of isolated and purified wild-type genomic DNA from human buccal cells. This control performed PCR utilizing published primers from Lago et al. (2017), named PWF and PWR that targeted the wild-type CFTR gene (Lago et al., 2017). An expected PCR product of 144 base pairs was regularly identified to support the integrity of wild-type CFTR gene.
By analyzing the CFTR gene, both wild-type and ΔF508-seeking primers were designed to work with a single common return primer (Table 1). Both of the allele-specific primers were designed with an intentional mismatch on the 3-most end in order to decrease non-specific binding (Simsek and Adnan, 2000). The authors designed the assay with goal to visualize amplified segment of approximately 1000 base pairs (ultimately was 989 bp).
Lastly, primers were designed to anneal to and thus detect the presence of wild-type adenovirus B7. These primers were tested in the presence of a sample of purified adenovirus B7 genomic DNA obtained from the laboratory of Dr. Adriana Kajon at the Lovelace Respiratory Research Institute (Metzgar et al., 2007).This
assay also initiated aimed to amplify approximately 1000 base pairs (ultimately was 1,027 bp). As a negative control, the primers were also tested against adenovirus E4 genomic DNA given they were not complementary to any section of the E4 DNA.
Optimal PCR conditions were determined by running multiple experimental trials while altering one variable at a time, such as annealing temperature, concentration of DNA, primer, and MgCl2. The lambda amplification was optimized with 1.5 mM MgCl2 and an annealing temperature to 56?. This provided adequate magnesium as a cofactor of Taq polymerase, as well as higher annealing temperature specificity in primer binding (Lorenz, 2012). The regular use and visualization of successful lambda amplification served as a positive control.
Control trials with the PWR and PWF primers from Lago et al. (2017) were successful with 1.5 mM MgCl2, and an annealing temperature of 59?, amplifying 144 base pairs of the CFTR gene. Obtaining successful results from the published primers served as demonstration of a successful isolation of purified DNA with intact CFTR gene.
The experimental diagnostic assay utilized designed primers tested in AS-PCR cocktails for both vs ΔF508-CFTR and wild-type CFTR.
The brightest bands of the positive controls analyzed supported approximately 1000 base pair amplification, very close to the target of 989 base pair amplification. Also, the negative controls did not contain any amplification products near 1k base pair band, suggesting the primers did not anneal to the mutant DNA in the target region, and all additional bands presented were likely a result of non-specific binding.
Lastly, the designed adenovirus-seeking primers were tested in PCR with both positive and negative templates and ultimately proved to be successful whereas approximately 1000 base pairs were amplified in the positive context. The negative context had no bands. These results support the demonstration of the ability of the B7-seeking primers to differentiate between different strains of adenovirus as designed. In further research, the adenovirus primers targeting B7 and E4 will be the primary focus and center of further investigation in order to further evaluate assays capable of distinguishing between the two strains. In order to develop this assay, a common forward primer on adenovirus E4 and B7 will be utilized and differing reverse primers will be designed that seek to anneal specifically to conserved ORF regions in the genome of the E4 strain or the B7 strain of adenovirus. The project will be pursued with the intention of assisting in testing alternative strategies for oligonucleotide primer-based medical diagnosis of viral genome within patients with cystic fibrosis.
CONFLICT OF INTERESTS
The authors have not declared any conflicts of interests.
ACKNOWLEDGMENTS
We would like to thank Drs. Andrew VanAlst, Matthew VanderPloeg and Allison Vlk for their assistance and scholarly discussions in the generation of this work. This work was funded by the Cystic Fibrosis Foundation and Pennsylvania Cystic Fibrosis Inc.
REFERENCES
|
Dooki MRE, Akhavan-Niaki H, Juibary AG (2011). Detecting common CFTR mutations by reverse dot blot hybridization method in cystic fibrosis first report from northern Iran. Iranian Journal of Pediatrics 21(1):51-57. |
|
|
Garibyan L, Avashia N (2014). Research techniques made simple: polymerase chain reaction (PCR). Journal of Investigative Dermatology 133(3):e6. |
|
|
Lago JEF, Cayarga AA, Gonza?lez YJG, Mesa TC (2017). A simple, fast, inexpensive method for mutation scanning of CFTR gene. BMC Medical Genetics 18(1):1-7. |
|
|
Lorenz TC (2012). Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. Journal of Visualized Experiments 63:e3998 |
|
|
Marson FA, Bertuzzo CS, Ribeiro MA, Ribeiro AF, Ribeiro JD (2013). Screening for F508del as a first step in the molecular diagnosis of cystic fibrosis. Jorno Brasileiro de Pneumologia 39:306-316. |
|
|
Metzgar D, Osuna M, Kajon AE, Hawksworth AW, Irvine M, Russell KL (2007). Abrupt emergence of diverse species B adenoviruses at US military recruit training centers. The Journal of Infectious Diseases 196(10):1465-1473. |
|
|
Miró-Cañís S, Capilla-Rubio S, Marzo-Checa L, Fontanals-Aymerich D, Sanfeliu-Sala I, Espasa-Soley M, Asensio-De-La-Cruz O (2017). Multiplex PCR reveals that viruses are more frequent than bacteria in children with cystic fibrosis. Journal of Clinical Virology 86:1-4. |
|
|
Mulot C, Stücker I, Clavel J, Beaune P, Loriot M (2005). Collection of human genomic DNA from buccal cells for genetics studies: comparison between cytobrush, mouthwash, and treated cards. Journal of Biomedicine and Biotechnology 3:291-296. |
|
|
Rafeeq MM, Murad HA (2017). Cystic fibrosis: current therapeutic targets and future approaches. Journal of Translational Medicine 15:1-9. |
|
|
Roqué M, Godoy CP, Castellanos M, Pusiol E, Mayorga LS (2001). Population screening of F508del, the most frequent mutation in the CFTR gene associated with cystic fibrosis in Argentina. Mutation in Brief 18(2):167-170. |
|
|
Simsek M, Adnan H (2000). Effect of single mismatches at 3'-end of primers on polymerase chain reaction. Journal for Scientific Research 2(1):11-14. |
|
|
Taylor A, Benedik M, Campbell A (1983). Location of the Rz gene in bacteriophage lambda. Gene 26(2-3):159-163. |
|
|
Thibodeau PH, Richardson III JM, Wang W, Millen L, Watson J, Mendoza JL, Du K, Fischman S, Senderowitz H, Lukacs, GL Kirk K, Thomas PJ (2010). The cystic fibrosis-causing mutation F508del affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. The Journal of Biological Chemistry 285:35825-35835. |
|
|
Xu W, Erdman DD (2001). Type?specific identification of human adenovirus 3, 7, and 21 by a multiplex PCR assay. Journal of Medical Virology 64(4):537-542. |
|
|
Yaku H, Yukimasa T, Nakano S, Sugimoto N, Oka H (2008). Design of allele?specific primers and detection of the human ABO genotyping to avoid the pseudopositive problem. Electrophoresis 20:4130-4140. |
|
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


