Full Length Research Paper

ABSTRACT
In Uganda and elsewhere in the world, tomato is an economically important vegetable and a good source of vitamins A and C. Being an important horticultural crop, its production is threatened by root-knot nematode infections that make plants susceptible to wilting, growth reduction and infection by other pathogens like bacteria and fungi. Identifying the root-knot nematode species on the crop is paramount in designing proper management strategies especially crop rotation and resistance. In this study, tomato roots infected with Meloidogyne spp. were collected from fields in Kyenjojo and Masaka districts in Uganda. Using perineal patterns and molecular diagnostics, the three most common Meloidogyne species were identified. Meloidogyne javanica omit and was the most common followed by Meloidogyne arenaria and Meloidogyne incognita.
Key words: Identification, root-knot nematodes, tomatoes, Uganda.
INTRODUCTION
In Uganda, tomato is an economically important vegetable mainly grown by small scale farmers (Tumwine, 1999). It is grown as a sole crop all year round with the major commercial varieties being Money maker, Marglobe, Henz and Roma (Tumwine et al., 2002). Nutritionally, tomato is a good source of vitamins A and C, with recent studies indicating lycopene which has antidioxidant properties to protect against cancer and heart diseases in red tomatoes (Rao and Agarwal, 2000). Tomato growing is one of the areas looked on for horticultural development in Uganda (Osiru et al., 2001) but yields per hectare are still very low, that is, 10 ton/ha (Ssekyewa, 2006). The Agricultural industry in Africa is at stake because of Meloidogyne populations, worsened by the fact that farmers have limited awareness or information on Meloigogyne spp in their gardens (Kagoda et al., 2010; Onkendi and Moleleki, 2013a). Heavy galling and rotting of roots on dying tomato plants infected by root-knot nematodes in the nine studied districts of Uganda was reported by Bafokuzara in 1996. Meloidogyne spp are considered to be the most wide spread and destructive plant-parasitic nematodes and can cause an estimated yield loss of 25 to 50% over wide areas of cultivated land (Taylor and Sasser, 1978). A loss of 24 to 38% has been reported in tomatoes (Sikora and Fernàndez, 2005). Speijer and Kajumba (2000) revealed that Meloidogyne spp and other plant parasitic nematodes cause banana yield losses of 50% in Uganda. So far, more than 100 species of root-knot nematodes have been described (Hunt and Handoo, 2009). Meloidogyne incognita, Meloidogyne javanica and Meloidogyne arenaria are the most common in the tropical regions while M. hapla, M. fallax and M. chitwoodi are successful in temperate and cooler regions (Karssen and Van Aelst, 2001). Under severe infections, plants wilt and die because of alterations in nutrient and water uptake (Kaloshian et al., 1996), an example of M. enterolobii on guava in Vietnam (Iwahori et al, 2009). Damage on plants is more pronounced in tropical climates than in temperate because of the favourable conditions for nematode survival and multiplication (De Waele and Elsen, 2007; Kaloshian et al., 1996).
Meloidogyne spp. lifecycle can take three weeks or some months depending on environmental factors such as availability of a suitable host, temperature and moisture (Taylor and Sasser, 1978). The infective second-stage juveniles move in the soil and penetrate the root tips of the host plant using their stylet (Karssen, 2002). The juveniles (J2) feed and undergo three molts (J3, J4 and adult). Sometimes, vermiform males develop and migrate out of the root while the females remain sedentary (Moens et al., 2009). Several management strategies, such as host plant resistance, destruction of residual crop roots, rotation with non-host plants, sanitation and avoidance, and careful use of nematicides, have been reported to effectively control root-knot nematodes (Barker and Koenning, 1998). However, the use of resistant varieties remains the most promising option, especially for small-scale farmers having limited resources. With species identification, the design of the above management strategies can be realized (Blok et al., 2002). This article present species of root-knot nematodes associated with tomatoes in the districts of Masaka and Kyenjojo in Uganda.
MATERIALS AND METHODS
Sample collection
Samples of tomato roots and soil infected with root-knot nematodes were collected from two districts; Kyenjojo in western Uganda (coordinates 00 37N, 30 37E) at an elevation of 1,400 m above sea level, and Masaka district in south western Uganda (coordinates 00 30S, 31 45E) at an average altitude of 1,115 m above sea level (Figure 1). For Kyenjojo, we collected samples from 3 villages; Katooke, Kinogero and Kyeniga and from Masaka district, samples were collected from two villages; Kyabakuza and Kimanya. Fruiting tomato plants were uprooted, and plants with galls were selected to constitute the samples. The whole rooting system of a plant and about 500 g of soil around the plant were collected and placed in a plastic bag, labeled carefully and kept in a refrigerator at 8°C until nematode extraction.
.png)
Nematode extraction and pure culture establishment
Nematodes were extracted from tomato roots with the Baermann funnel technique (Hooper, 1990). Therefore, roots were washed in running water, chopped into small pieces and placed on a filter paper (Ederol Rundfilter, 40 g/m2, Munktell Filter AB, Falun, Sweden) lined in a 2 mm sieve. The sieves were then placed on glass funnels filled with water to a level that just covered the bottom of the sieve and roots. The stem of the funnels connected to a rubber tube was closed with two metal clips. Second-stage juveniles hatched, moved through the filter paper and concentrated at the bottom of the rubber tube. Water was daily added to the funnels to prevent drying of roots as water evaporated. To culture the nematodes, newly hatched J2s tapped from Baermann funnels were inoculated on 3 weeks old tomatoes (Solanum lycopersicon cv. Marmande) that were earlier transplanted in 17cm diam. pots filled with sterilized soil (100°C, 16 h). Plants were maintained at 22 to 28°C in the green house with a 14 h light period.
After 6 weeks, infected tomato plants were removed carefully from their pots and the roots were washed to remove excess soil. Individual egg masses were picked at random using a stereoscopic microscope and made to propagate on tomato cv. Marmande to obtain pure cultures. Plants were kept in the green house with the same conditions mentioned previously for 6 weeks.
Collection of materials for perineal patterns and molecular identification
Infected roots from pure cultures were collected and washed. Adult females for perineal patterns were carefully teased out of the roots and kept in a solution of 0.9% sodium chloride. The rest of the root systems were put on Baermann funnels to collect juveniles and males for DNA analysis.
Identification
Perineal patterns
Perineal patterns were prepared according to a method described by Taylor and Netscher, (1974). Females were teased out of the root and placed in a solution of 0.9% sodium chloride. For each pure culture, five perineal patterns were cut from live egg-laying females in 45% lactic acid and mounted in glycerin. They were viewed under a light microscope (magnification1000×), and pictures were taken using a biological imaging soft-ware (cell D).
DNA extraction
DNA of J2s isolated from roots was extracted based on Holterman et al., (2006). Therefore, single to five J2s were picked up from water that was tapped from the funnels using a picking needle and added to 25 µL sterile water in a 0.2 ml-PCR tube. 25µL of worm lysis buffer containing 0.2M Nacl, 0.2M Tris-Hcl (Ph8.0), 1% (v/v) beta-mercaptoethanol and 800 µg/ml Proteinase K was added. The samples were then incubated for 1 h and 30 min at 65°C followed by 5 min. incubation at 99°C was in a thermcycler. The DNA was immediately stored at -20°C waiting further analysis.
PCR amplification
Primers used for DNA amplification are given in Table 1. The quality of the extracted DNA was checked using a universal primer D2a and D3b (De Ley et al., 1999), amplifying the D2 and D3 expansion region of the 28S rDNA nuclear gene. Positive samples with the D2a and D3b primers were further amplified using species-specific primers for M. incognita, M. javanica and M. arenaria (Zijlstra et al., 2000) based on the given specifications. The SCAR amplification reactions were performed in 0.2 ml PCR tubes with the reaction volumes of 50 µL. A master mix containing 34.6 µL double distilled water, 5 µL10 x PCR buffer, 6 µL MgCl2, 1 µL of dNTPs mix, 0.5 µL reverse primers, 0.5 µL forward primers and 0.4 µL Taq DNA polymerase (Phamarcia) was prepared. Each volume was multiplied by the number of samples to be analyzed and brought into a 1ml tube and vortexed. Finally 48 µL of the master mix was pipetted into 0.2 ml Eppendorf tubes containing 2 µL of DNA. The tubes were then transferred to a thermcycler and the cycles ran according to (Zijlstra et al., 2000).The PCR products were then used immediately or kept at -20°C until use.
Mitochondrial DNA analysis
The primers C2F3 and 1108 (Powers and Harris, 1993) were used to amplify the mtDNA region between the cytochrome oxidase sub unit II (COII) and the 16S rRNA genes. These primers were used to further confirm the results of the species-specific primers. Amplifications were done in 50 µL reaction volumes. A master mix containing 34.6 µL ddH2O, 5 µL 10x PCR buffer, 6 µL MgCl2, 1 µL of dNTPs mix, 0.5 µL reverse primers, 0.5 µL forward primers and 0.4 µL Taq DNA polymerase (Phamarcia) was prepared. Finally 48 µL of the master mix were pipetted plus 2 µL of DNA into a 0.2ml tube. The temperature for the reaction was 94°C for 2 min. followed by 35 cycles of 94°C for 30 s, 50°C for 30 s and 72°C for 2 min. The final elongation followed at 72°C for 8 min. JMV primers (Wishart et al., 2002), were used to detect M. hapla, M. chitwoodi and M. enterolobii.
Electrophoresis
The reaction products were resolved by electrophoresis on a 1.5% agarose gel in 1 × Tris borate EDTA buffer. For each PCR sample, 1 µL loading buffer-blue orange x6 was added to 5 µL PCR product in 0.2 ml Eppendorf tubes. The mixture was then added to the gel wells. A DNA marker (ladder 100 bp or 1 kb) was added into the first and the last well of gels. Electrophoresis was run at 100 v for 40 min. The gels were then stained in ethidium bromide bath for 30 min Visualization of gels was done on a UV- trans-illuminator and the picture taken.
RESULTS
Pure culture establishment
A total of 30 egg masses were collected from tomato plants to set up pure cultures. From these, 24 egg masses were successfully cultured and used for species identification.
Identification
Using molecular and perineal patterns methods of identification, M. javanica, M. arenaria and M. incognita was identified (Table 2). Surprisingly one of the cultures had a mixture of M. javanica and M. arenaria, and some of the cultures remained unidentified.
Perinneal patterns
Meloidogyne javanica
The perineal patterns (Figure 2) were typical for M. javanica with a rounded to flattened dorsal arch and conspicuous lateral lines that clearly separated the dorsal and ventral regions of the patterns.

Meloidogyne arenaria
The M. arenaria females’ perineal patterns had the characteristic rounded to flattened, low dorsal arch, with some of the striae forming a small shoulder marked by bifurcations and curved to close striae in the lateral field and occasionally with a slight lateral line (Figure 3).

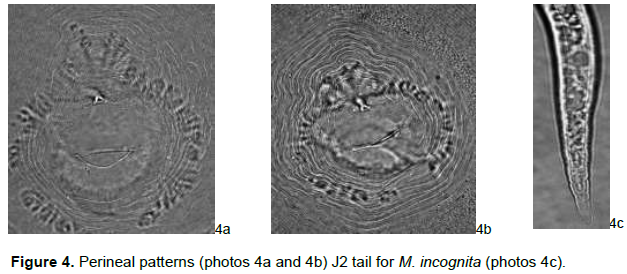
Meloidogyne incognita
Females showed characteristic oval to rounded perineal patterns with a high dorsal arch and wavy striae which bend towards the lateral lines and the absence of distinct lateral line incisures typical of this species (Figure 4).
Molecular identification
Ribosomal DNA amplification with D2A D3B primers
DNA obtained from single to five J2s from the 24 cultures was used. The D2D3 extension fragment of the 28S rDNA was used in order to check the quality of DNA and prevent false negatives in the species-specific PCRs. The samples checked showed the correct band size of 750 bp (data not shown).
PCR amplification of sequence characterized amplified region (SCAR)
The SCAR primer pairs (Table 1) were used for the diagnosis of M. incognita, M. javanica and M. arenaria.
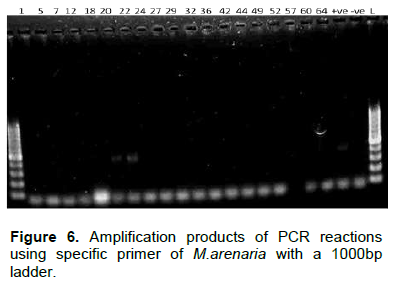
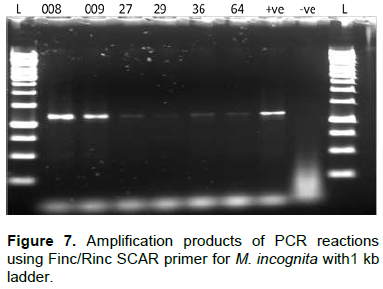
PCR amplification of COII/16SrRNA of the mitochondrial DNA
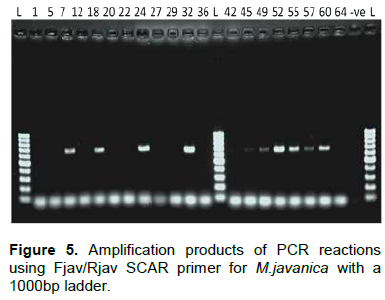
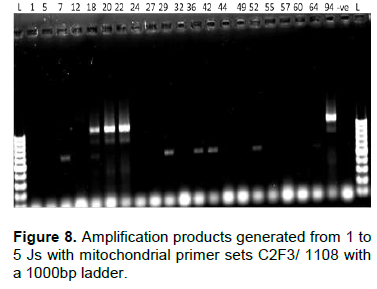
The primer set C2F3 and 1108 was used to amplify the COII/16S rRNA gene of the mitochondrial DNA. It clearly amplified M. arenaria cultures with a base pair product of 1100 bp, but did not amplify for M. javanica and M. incognita as expected. On the same gel, weak bands of around 600 bp were detected for samples Ktk5, Ktk4, Ky107, Ky106, Kbz1 and Hr103 (Figure 8). To check if these bands could be allocated to M. chitwoodi or M. hapla, we used a one step multiplex PCR using JMV primers (table 1). Neither M. chitwoodi, M. hapla nor M. fallax were detected. To detect the presence of M. enterolobii in our samples, primers JMV1 and JMV tropical were used. An amplification product of 615 bp in all the tested samples indicated the absence of M. enterolobi.
DISCUSSION
Accurate and careful identification of Meloidogyne species infecting crops is a core for efficient use of plant resistance and successful management of root-knot nematodes. The severe infections on tomato plants and growth impairment observed in fields during sampling, requires immediate attention and implementation of feasible control strategies. The purpose of this study was to identify root-knot nematode species found in the districts of Kyenjojo and Masaka in Uganda. This gave light on the Meloidogyne species present in the areas. Results from the sampled areas indicate the presence of M. incognita, M. arenaria and M. javanica which is not a surprise as they are mentioned as the most common Meloidogyne species in tropical regions (Taylor and Sasser, 1978; Moens et al., 2009) like Uganda where annual temperatures are between 15 to 30°C. M. javanica dominated the presently studied samples with 45%, followed by 37.5% of unidentified species, 12.5% M. arenaria and lastly M. incognita with 4%. A similar finding was reported by Nono-womdin et al., (2002) in Tanzania and Naz et al., (2012) in Pakistan where M. javanica was widely distributed than M. incognita. These results are in disagreement with Eisenback et al. (1981) where M. incognita was the most prevalent among all Meloidogyne species in the studied areas of the international Meloidogyne project. As reported by Oliveira et al. (2011), the undescribed species in the samples was expected with the present movement of plant-derived materials around the world, climate change, continuing growth of human population and consequent changes in land use and agricultural practices. There are also high possibilities of new species being introduced into the continent because of limited quarantine regulations. These come with implications on the agricultural sector like reduced crop yields, high costs of control and loss of revenue. Perineal patterns are considered important for differentiating Meloidogyne species. However, in this study, they were found to be variable making it difficult to separate species based on them alone. This was evident in some perineal patterns of M. arenaria, M. incognita and the species not identified having visible lateral lines and could easily be confused to be patterns of M. javanica. The same confusion was reported by Rammah and Hirschmann (1990), García and Sánchez- Puerta (2012).
SCAR primer sets were used to detect tropical Meloidogyne species; Fjav/Rjav, for M. javanica, Finc/Rinc for M. incognita and Far/Rar primers for M. arenaria successfully gave PCR products of 670 bp, 1200 bp and 420 bp, respectively. These results showed agreement with earlier studies (Zijlstra et al., 2000; Adam et al., 2007). However, Finc/Rinc for M. incognita formed other weak amplification products of the same length with the positive control. This suggeststhat probably, the species had some relationship with M. incognita though female perineal patterns which did not match exactly. Failure of the primer to give a strong band in the other isolates could also have been because of low copy in the amounts of the corresponding targeted regions of the primer sets (Adam et al., 2007). Moreover, according to literature studied, no information has been given in regard to such behavior of the primer.
The mitochondrial DNA is a good source of genetic markers for species and population genetics identification (Blok and Powers, 2009). It is important in molecular phylogenetic and diagnostic studies of root-knot nematodes (Stanton et al., 1997) because of its high copy number and fast evolution rate (Moritz et al., 1987). The primers C2F3/1108 were previously tested and reported as effective for amplifying the COII/LrRNA region of mtDNA for M. javanica, M. arenaria and M. incognita (Powers et al., 2005; Blok et al., 2002). For this study, they only gave an amplification product of 1.1 kb for M. arenaria. The primer did not give amplification products for M. incognita and M. javanica as it was expected. The M. arenaria result was in agreement with that of Blok et al. (2002) and Powers and Harris (1993). With the same primer, an amplification product of approximately 600 bp was observed. Such product size is close to the products given by M. chitwoodi and M. hapla using the same primer. JMV primers (Wishart et al., 2002) were used to specifically detect M. hapla and M. chitwoodi but no result was given. Moreover, depending on the annual temperatures of the sampled areas, the presence of these two species is not expected because M. hapla and M. chitiwoodi are found in cooler climates. Since Powers and Harris (1993) tested the primers for North American species, the general applicability of the primer on all Meloidogyne species was not confirmed. This gives some evidence to suggest that the amplification product could be of a new species outside the ones studied. Amplification for other species (M. incognita and M. javanica) failed, possibly due to inconsistent PCR conditions. Similar abnormalities of the primer were reported by Devran and SöÄŸüt (2009) where the primer did not give any result. According to Adam et al. (2007), the inability to identify the species in some samples could be as a result of changes in priming sites that prevent amplification of the diagnostic product and changes in regions between the primer sites leading to the formation of products different from what is usual or the samples contain species different from the tested ones.
The use of species-specific primers was very helpful and gave confidence in the identification process. However, since the primers are species-specific for M. incognita, M. javanica and M. arenaria, they could not amplify other nematode species outside that category.
To assist in the identification of Meloidogyne species, the use of RAPD technique needs to be considered. Already in use today, random amplified polymorphic DNA are useful in characterizing intra and interspecific variation since there is no need for prior information on the DNA sequence of the targeted genome (Block and Powers, 2009). For example, Adam et al. (2007) found consistent amplification patterns from Males, females, and J2 s of M. javanica using RAPDs. Cenis (1993) reported the usefulness of RAPD-PCR to diagnose and address the unsolved questions on genetic variation and population genetics of root-knot nematodes. Furthermore, sequence analysis of rDNA has increased in importance in the identification of Meloidogyne spp. Different sets of primers have been reported to amplify different regions of rDNA. For example, the primers designed by Vrian et al. (1992) have been used to amplify the ITS region for Meloidogyne spp. to form a product of approximately 800-bp which can then be sequenced to produce species-specific primers or enzyme digested fragments (Blok and Powers, 2009). Blok and Powers, (2009) also reported that species that are difficult to separate using morphological and biological features like M. hispanica from M. arenaria and M. incognita have been separated by the comparisons of their ITS, 18S and D2/D3 a region within the 28S gene. Sequence polymorphisms in the IGS region have been used by Wishart et al. (2002) and Blok et al. (2002) to distinguish M. chitwoodi and M. fallax from other species like M. enterolobii, M. hapla and M. incognita / M. arenaria / M. javanica using JMV primers.
A mixture of two species in a culture presumed to be from a single egg mass which could have been caused by two females lying close together in the root and forming an egg mass that can easily be mistaken to be from one female. The same finding was reported by Devran and SöÄŸüt, (2009). Further research should employ other methods like use of sequence-based methods on the ribosomal DNA, mitochondrial DNA, and use of isozymes to describe species that were not identified.
CONCLUSION
There are new emerging species of root not nematodes and it is difficult to conclude that a single method is sufficient to delineate the species. Generally, there is limited information on the species of root-knot nematodes and the associated yield losses in Africa. Identification of these nematodes will guide management programs because some species are host specific.
CONFLICT OF INTERESTS
The authors have not declared any conflict of interests.
ACKNOWLEDGEMENTS
With great pleasure I extend my sincere gratitude to the Belgian government through VLIR-UOS scholarships for the financial support. Many thanks to the institute of agricultural and fisheries research (ILVO) for providing unlimited facilities to run the research.
REFERENCES
|
Adam M, Phillips M, Blok V (2007). Molecular diagnostic key for identification of single juveniles of seven common and economically important species of rootâ€knot nematode (Meloidogyne spp.). Plant Pathol. 56:190-197. |
|
|
Bafokuzara ND (1996). Incidence of different nematodes on vegetable and fruit crops and preliminary assessment of yield loss due to Meloidogyne species in Uganda. Nematol. Bras. 20:32-43. |
|
|
Barker KR, Koenning SR (1998). Developing sustainable systems for nematode management. Ann. Rev. Phytopathol. 36:165-205. |
|
|
Blok VC, Powers TO (2009). Biochemical and molecular identification. In R. Perry,M. Moens, and J. Starr, (eds). Root-Knot Nematodes. CABI Publishing, Wallingford, UK. pp. 98-118. |
|
|
Blok VC, Wishart J, Fargette M, Berthier K, Phillips MS (2002). Mitochondrial DNA differences distinguishing Meloidogyne mayaguensis from the major species of tropical root-knot nematodes. Nematology 4:773-781. |
|
|
Cenis JL (1993). Identification of four major Meloidogyne spp. by Random Amplified Polymorphic DNA (RAPD-PCR). Phytopathol.NY. Baltimore then st Paul 83:76-76. |
|
|
De Ley P, Felix MA, Frisse LM, Nadler SA, Sternberg PW, Thomas WK (1999). Molecular and morphological characterisation of two reproductively isolated species with mirror-image anatomy (Nematoda: Cephalobidae). Nematology 1:591-612. |
|
|
De Waele D, Elsen A (2007). Challenges in tropical plant nematology. Ann. Rev. Phytopathol. 45:457-485. |
|
|
Devran Z, SöÄŸüt MA (2009). Distribution and identification of root-knot nematode from Turkey. J. Nematol. 41:128-133. |
|
|
Eisenback JD, Hirschmann H, Sasser JN, Triantaphyllou AC (1981). A guide to the four most common species of root-knot nematodes (Meloidogyne species). A Cooperative Publication of the Departments of Plant Pathology and Genetics, North Carolina State University and the United States Agency for International Development: Raleigh, NC. P. 4. |
|
|
García LE, Sánchez-Puerta MV (2012). Characterization of a root-knot nematode population of Meloidogyne arenaria from Tupungato (Mendoza, Argentina). J. Nematol. 44(3):291. |
|
|
Holterman M, Van der Wurff A, van den Elsen S, van Megen H, Bongers T, Holovachov O, Bakker J, Helder J (2006). Phylum-wide analysis of SSU RDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades. Mol. Biol. Evol. 23:1792-1800. |
|
|
Hooper D (1990). Extraction and processing of plant and soil nematodes. In Luc L, Sikora RA, Bridge J (eds). Plant parasitic nematodes in subtropical and tropical agriculture. CAB International, Wallingford, UK. pp. 45-68. |
|
|
Hunt DJ, Handoo ZA (2009). Taxonomy, identification and principal species. In Perry RN, Moens M, Starr JR (eds.). Root-knot nematodes CABI Publishing, UK. pp. 55-97. |
|
|
Iwahori H, Truc NTN, Ban DV, Ichinose K (2009). First report of root-knot nematode Meloidogyne enterolobii on guava in Vietnam. Plant Dis. 93(6):675-675. |
|
|
Kagoda F, Derera J, Tongoona P, Coyne DL (2010). Awareness of plant parasitic nematodes, and preferred maize varieties, among smallholder farmers in East and Southern Uganda: implications for assessing nematode resistance breeding needs in African maize. Int. J. Pest. Manage. 56:217-222. |
|
|
Kaloshian I, Williamson V, Miyao G, Lawn D, Westerdahl B (1996). "Resistance breaking" nematodes identified in California tomatoes. California Agric. 50:18-19. |
|
|
Karssen G (2002). The plant-parasitic nematode genus Meloidogyne Göldi, 1892 (Tylenchida) in Europe. Leiden, Brill Academic Pub. 160 p. |
|
|
Karssen G, Moens M, Perry R (2006). Root-knot nematodes. In Perry RN, and Moens M. (eds.). Plant. Nematol. CABI Publishing, Wallingford, UK. pp. 59-90. |
|
|
Karssen G,Van Aelst AC (2001). Root-knot nematode perineal pattern development: a reconsideration. J. Nematol. 3:95-111. |
|
|
Moens M, Perry RN, Starr JL (2009). Meloidogyne species a diverse group of novel and important plant parasites. In Perry RN, Moens M, Starr JL(eds.). Root-knot nematodes CABI International, Cambrige, MA (USA), pp. 1-17. |
|
|
Moritz C, Dowling T, Brown W (1987). Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Ann. Rev. Ecol. Syst. 18:269-292. |
|
|
Naz I, Palomares-rius JE, Blok V, Ali S, Ahmed M (2012). Prevalence, incidence and molecular identification of root-knot nematodes of tomato in Pakistan. In an International Conference on climate Change: A challenge for Agriculturists. 28-30, May, 2012, the University of Agriculture, Peshawar (Local) 11(100):16546-16556. |
|
|
Nono-Womdim R, Swai I, Mrosso L, Chadha M, Ope-a R (2002). Identification of root- knot nematode species occurring on tomatoes in Tanzania and resistant lines for their control. Plant Dis. 86:127-130. |
|
|
Oliveira CMGD, Monteiro AR, Blok VC (2011). Morphological and molecular diagnostics for plant-parasitic nematodes: working together to get the identification done. Trop. Plant Pathol. 36:65-73. |
|
|
Onkendi EM, Moleleki LN (2013a). Distribution and genetic diversity of root-knot nematodes (Meloidogyne spp.) in potatoes from South Africa. Plant Pathol. 62:1184-1192. |
|
|
Osiru M, Rubaihayo P, Opio A (2001). Inheritance of resistance to tomato bacterial wilt and its implication for potato improvement in Uganda. Afr. Crop Sci. J. 9:9-16. |
|
|
Powers TO, Harris T (1993). A polymerase chain reaction method for identification of five major Meloidogyne species. J. Nematol. 25:1-6. |
|
|
Powers TO, Mullin P, Harris T, Sutton L, Higgins R (2005). Incorporating molecular identification of Meloidogyne spp. into a large-scale regional nematode survey. J. Nematol. 37:226-35. |
|
|
Rammah A, Hirschmann H (1990). Morphological comparison of three host races of Meloidogyne javanica. J. Nematol. 22:56-68. |
|
|
Rao AV, Agarwal S (2000). Role of antioxidant lycopene in cancer and heart disease. J. Am. Coll. Nutr. 19:563-569. |
|
|
Sikora RA, Fernandez E (2005). 9 nematode parasites of vegetables. In Luc M, Sikora RA, Bridge J (Eds.), Plant parasitic nematodes in subtropical and tropical agriculture, second ed. CAB International, Wallingford, Oxford, UK. pp. 319-392. |
|
|
Speijer PR, Kajumba C (2000). Yield loss from plant parasitic nematodes in East African highland banana (Musa spp. AAA). Acta Hortic. 540:453-459. |
|
|
Ssekyewa C (2006). Incidence, distribution and characteristics of major tomato leaf curl and mosaic virus diseases in Uganda. PhD thesis, Ghent University, Ghent, Belgium 233 p. |
|
|
Stanton J, Jugall A, Moritz C (1997). Nucleotide polymorphisms and an improved PCR based mtDNA diagnostic for parthenogenetic root-knot nematodes (Meloidogyne spp.). Fund. Appl. Nematol. 20:261-268. |
|
|
Taylor A, Sasser J (1978). Biology, identification and control of root-knot nematodes North Carolina State University Graphics, Raleigh. |
|
|
Taylor DP, Netscher C (1974). An improved technique for preparing perineal patterns of Meloidogyne spp. Nematology 20:268-269. |
|
|
Tumwine J (1999). Towards the development of integrated cultural control of tomato late blight (Phytophthora infestans) in Uganda. PhD Thesis, Department of Phytopathology, Wageningen Agricultural University, The Netherlands 152 p. |
|
|
Tumwine J, Frinking H, Jeger M (2002). Tomato late blight (Phytophthora infestans) in Uganda. Int. J. Pest. Manage. 48:59-64. |
|
|
Vrain TC, Wakarchuk DA, Levesque AC, Hamilton RI (1992). Intraspecific RDNA restriction fragment length polymorphism in the Xiphinema americanum group. Fund. Appl. Nematol. 15:563-573. |
|
|
Wishart J, Phillips MS, Blok VC (2002). Ribosomal intergenic spacer: a polymerase chain reaction diagnostic for Meloidogyne chitwoodi fallax M, hapla M. Phytopathology 92(8):884-892. |
|
|
Zijlstra C, Donkers-Venne DTHM, Fargette M (2000). Identification of Meloidogyne incognita, javanica M, M arenaria using sequence characterised amplified region (SCAR) based PCR assays. Nematology 2:847-853. |
|
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


