Full Length Research Paper

ABSTRACT
Saccharomyces cerevisiae has two hexokinases ScHxk1 and ScHxk2 that catalyze ATP-dependent phosphorylation of glucose and other hexoses. ScHxk2 plays an important role in glucose metabolism and the process of bioethanol production. The presence of xylose in the fermentation medium was found to inhibit ScHxk2. Therefore development of ScHxk2 variants that are resistant to the action of xylose is needed. In the current study, in-silico investigation was done aiming to select the amino acids in ScHxk2 that can be targeted in an engineering experiment. Using Autodock Vina, xylose binding to ScHxk2 structure (PDB 1IG8) was predicted. The information available about hexokinase family in the publicly available hexokinase 3DM database were investigated and the conservancy patterns for potential residues in the xylose-binding site were extracted. The study eventually presented 54 suggested mutants that might lead to a xylose-tolerant hexokinase. Top correlated positions in the hexokinase superfamily indicated 6 proposed double-mutants that are worth to be included in the proposed smart library.
Key words: Library design, hexokinase, 3DM, database, protein engineering, xylose.
INTRODUCTION
Saccharomyces cerevisiae hexokinase 2 (ScHxk2) catalyzes the phosphorylation of glucose to glucose 6-phosphate via transfer of an ATP phosphate group to the 6-position on the glucose. In addition to this catalytic activity, ScHxk2 is involved in the regulation of other genes using glucose catabolite repression mechanism (Moreno and Herrero, 2002). ScHxk2 is found to be irreversibly inhibited by xylose. Xylose has similar structure to glucose and has the ability to bind to the glucose binding site in ScHxk2 structure. In the presence of ATP, xylose was found to induce autophosphorylation of the ScHxk2 at Ser158 position (Heidrich et al., 1997). Therefore S. cerevisiae cannot efficiently utilize glucose in the presence of xylose. Xylose is an abundant hydrolytic product obtained from the pre-treatment of the lignocellulosic material used as feedstock for bioethanol production. Therefore, development of ScHxk2 variants able to efficiently utilize glucose in the presence of high concentration of xylose is required.
The directed evolution approach to engineer enzyme variants with new properties has provided many successes over the last two decades (Wang et al., 2012). This approach necessitates the creation of a library of thousands of mutants followed by use of a good screening system to select for the desired variants. For efficient directed evolution experiment, the library size should be reduced to ease the screening effort. With the aid of advanced computer programs, the design of the library for directed evolution experiment becomes smarter and the library size becomes smaller (Nobili et al., 2013; Wijma and Janssen, 2013).
The 3DM database systems are high quality structural alignments of related protein structures (Joosten, 2007; Kuipers et al., 2010b). For each group of related protein structures, a superfamily is built and a consensus core is assigned. The consensus facilitates a unified numbering scheme for all the sequences in the superfamily that is, the 3DM-numbering scheme. This 3DM numbering allows knowledge transfer between similar residues that occupy the same spatial position in homologous protein structures. The 3DM also collects mutation information found in literature and link them to the 3DM numbers (Kuipers et al., 2010a). The successful application of 3DM for enzyme engineering, either for improving thermostability or catalytic property such as enantioselectivity has been described in Cerdobbel et al. (2011); Jochens et al. (2010) and Nobili et al. 2013).
In the current report, investigation of the ScHxk2 structure within the light of 3DM information was done. Selected positions in the active site were evaluated and suggested mutations were given that might increase the enzyme resistance to the inhibitory effect of xylose.
MATERIALS AND METHODS
Structure analysis of ScHxk2 was based on the protein structure deposited in the protein data bank PDB 1IG8 (Kuser et al. 2000), using YASARA Structure software (Krieger et al. 2002). The docking experiment of xylose was done using AutoDock Vina integrated in YASARA Structure software (ver. 13.9.8). Xylose coordinates, were extracted from the PDB: 2E2Q and energy was minimized using YASARA Structure and AMBER99 force field (Trott and Olson, 2010). The analysis of contacts between xylose and the surrounding residues in 1IG8 after the docking experiment was done using the Analysis/Contact function in YASARA Structure.
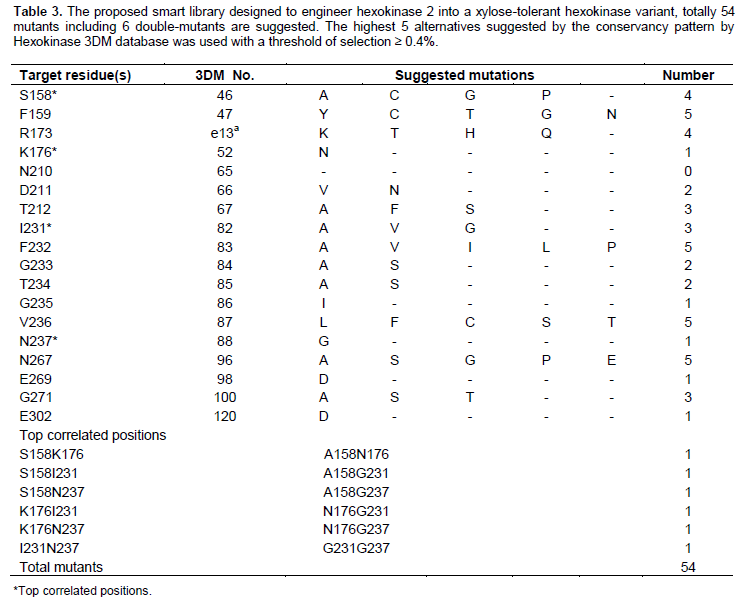
The amino acid sequence of ScHxk2 [UniProt accession no. P04807] was searched in the 3DM hexokinase database and was found under the identifier name: HXKB_YEAST. ScHxk2 belongs to the subfamily 1IG8A that includes 97 aligned sequences, and the consensus core contains 19 variable regions. The 3DM hexokinase database is built based on 20 structures, 547 aligned sequence and includes information from 2177 mutations. The superfamily has a consensus core of 214 residues. The superfamily is divided into 5 subfamilies based on five prototype protein data bank (PDB) structures namely: 1BDGA, 1IG8A, 1SZ2B, 2DGKN and 3CZAN. Each of these subfamilies has subfamily consensus which has some minor differences compared to the superfamily consensus. 3DM conservancy pattern for residues identified as targets for mutations were extracted, and the library designed considered the highest five alternatives for each target residues with a threshold of selection ≥ 0.4%.
RESULTS AND DISCUSSION
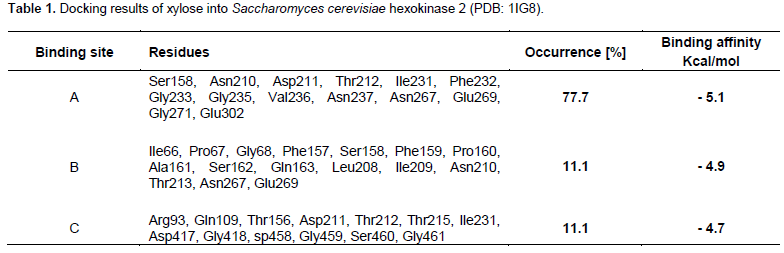
S. cerevisiae has two isoenzymes of hexokinase that catalyse phosphorylation of glucose in addition to other hexoses e.g. fructose and mannose. ScHxk2 structure has been determined using X-ray diffraction at a resolution of 2.2Å (Kuser et al., 2000). The structure has been determined without co-crystalized substrates or inhibitors. 1IG8 is composed of two domains that show significant movement upon glucose binding. Therefore, 1IG8 has two conformations referred to as either open or closed conformation. The docking experiment performed in the current report using AutoDock Vina, showed that xylose could probably occupy three main different binding sites inside 1IG8 structure (Table 1). The binding site A showed higher occurrence rate (77.7%) and slightly better binding affinity (-5.1 kcal/mol) compared to the other sites B and C (Table 1). Kuser et al. (2000) defined the glucose binding site based on the obtained X-ray crystallographic and modeling data. They found glucose to be coordinated by extensive hydrogen bonding with the surrounding polar residues. Figure 1 shows different poses of xylose docked into the binding site A. The highly conserved residues: Asp211, Glu302, and Asn237 are within 5-Å distances to xylose (Figure 1). Xylose was X-ray determined in a hexokinase derived from the hyperthermophilic archaeon Sulfolobus tokodaii at 2.0Å resolution (PDB: 2E2Q) (Nishimasu et al., 2007). Xylose was found coordinated by three acidic residues: Asp140, Asp95 and Asp71 in 2E2Q active site. These acidic residues are the equivalent to the residues Glu203 and Asp211 found in binding site A in 1IG8 (Figure 1).
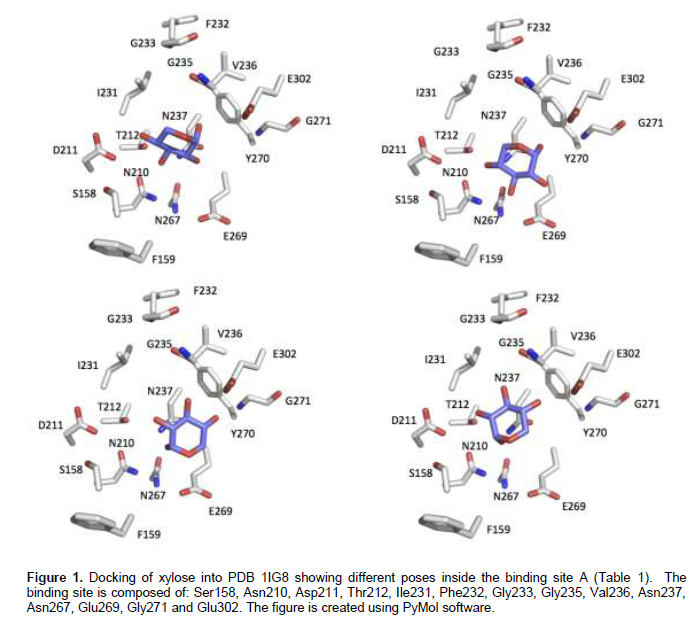
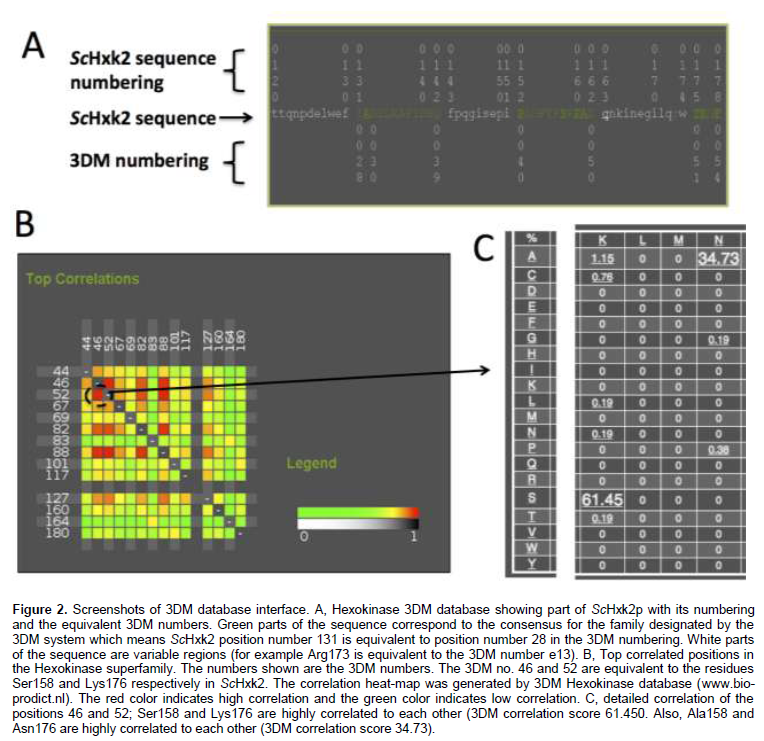
The basic idea in 3DM database is the numbering scheme that allows identification of equivalent amino acid residues in the space for a certain family of proteins. Figure 2 shows screenshots of hexokinase 3DM database interface. Figure 2A shows ScHxk2 sequence with two numbering schemes: the original sequence numbering and 3DM numbering. The green parts of the sequence belong to consensus core of the superfamily and the white parts belong to variable regions. Figure 2B shows the top correlated positions in the hexokinase superfamily and Figure 2C shows the detailed correlation score given by the 3DM to the correlated positions no. 46 and 52. Table 2 shows the residues of ScHxk2 selected as potential targets for mutations. The residues are based on the docking results of xylose and information in literature regarding the glucose binding inside related hexokinases. One aspect of smart library design is to exclude whatever mutations might be deleterious to the protein proper folding. The 3DM was consulted regarding the selected targeted mutation residues (Table 2). The conservancy pattern for each of targeted residues was extracted from the 3DM and presented as percentages in Table 2. It was observed that some target residues were highly conserved such as Asn210 that has been found to have 99.27% predominance in the hexokinase super-family. Such a high percentage does not recommend mutation of such position and therefore was excluded from the proposed library (Table 3). Bergdahl et al. (2013) engineered ScHxk2 targeting selected residues in the glucose binding site. Comparing our library (Tables 3) to the results obtained by Bergdahl et al., showed interesting observations. First, there is an agreement of the successful mutation described by the Bergdahl et al. (2013) to the 3DM conservancy pattern that is, Phe159Tyr. Phe159Tyr corresponds to the 3DM number 47 and Tyr is found in 9% of the total sequences of the superfamily (Table 2), which is a relatively high percentage. More interestingly, when the conservancy is calculated based on the subfamily 1IG8A, Tyr is found in 47% of all the sequences at this position (3DM no 47). This mutation specifically agrees with the 3DM suggestion. In another example that shows that statistical data might give suggestions that do not match with conventional mutation strategies, 3DM has not suggested to replace the basic residue Lys176 (3DM number 52) with its related residue Arg at any instance (Table 2), instead Asn was found to be the strong candidate for a mutation (34 %). Similarly, the residue Asp211 (3DM no. 66) has never been found to be replaced by Glu residue. Intriguingly, Arg was found at this site in a small percentage (0.2 %) of sequences. The Gly271 residue (3DM no. 100) was found to be replaced by Ser, Thr and Ala residues at probabilities 3.5, 2.6 and 1.5% respectively, which are much higher than the Cys residue (0.4%) chosen by Bergdahl et al. (2013) in their engineering experiments. Similarly, the strong candidate to replace the position Thr212 (3DM no. 67) is Phe (31.4%) and not Ser (Bergdahl et al., 2013).
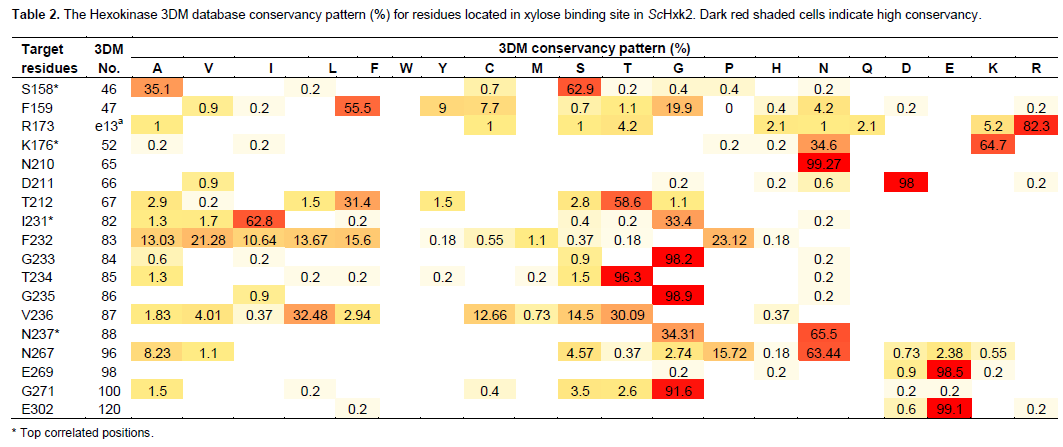
A very important feature that is provided by the 3DM database is the identification of the correlated positions. Correlated positions within a superfamily mean that a residue is found in nature to mutate simultaneously in correlation with another residue. This kind of correlated mutations might be of importance for correct protein folding or certain catalytic function. The correlated positions, however, are almost impossible to using the computational design protocols. Figures 1B and 1C show the top correlated positions in the hexokinase superfamily. The correlation analysis of the superfamily show that there are strong correlations between the positions 46 and 52 which are equivalent to the ScHxk2 positions Ser158 and Lys176 respectively. Ser158 has been defined as the site of phosphorylation upon xylose binding in the presence of ATP resulting in inactive hexokinase. Mutation of this residue to Ala, Cys, Gly, or Pro residues will lead to proper folded proteins according to the 3DM information (Table 2). However, this position is annotated as highly correlated to other positions in the hexokinase superfamily namely the 3DM positions 52, 82 and 88. Therefore, we suggested mutating this position to be in accordance with the correlation data provided by 3DM (Table 3). If the3DM position 46 (ScHxk2 Ser158) is mutated to Ala the 3DM position 52 (ScHxk2 Lys176) is mutated to Asn (3DM correlation score 34.37). This correlation was observed in 184 sequences all of which are under the subfamily 1SZ2B. Heidrich et al. (1997) mutated Ser158 to Ala and the activity was dramatically decreased compared to the wild type. The correlation of these two positions might interpret the recorded decreased enzymatic activity. Lys176 and Asn176 are found to interact by hydrogen bonding to the glucose in the active site as determined by X-ray experiments in the PDB entries: 1SZ2 and 1BDG. Therefore, the pairs Ser158Lys176 or Ala158Asn176 contribute to proper binding of glucose in the active site and have strong influence on the enzymatic activity.
It can be concluded from the present investigation that statistical information available for the hexokinase 3DM superfamily can be efficiently exploited to engineer the yeast hexokinase 2. A library of a very small size is prepared that can be easily implemented in the laboratory. Table 3 shows a list of positions in ScHxk2 active site and the most probable point mutations that can be experimentally done according to 3DM consensus. We envisage that the application of the proposed mutations can lead to deeper understanding of the structure-function relationship of ScHxk2 and identification of new enzyme variants that might resist the inhibitory action of xylose.
CONFLICT OF INTERESTS
The authors have not declared any conflict of interests.
REFERENCES
|
Bergdahl B, Sandström AG, Borgström C, Boonyawan T, van Niel EWJ, Gorwa-Grauslund MF (2013). Engineering yeast hexokinase 2 for improved tolerance toward xylose-induced inactivation. PloS one 8(9):e75055-e75055. |
|
|
Cerdobbel A, De Winter K, Aerts D, Kuipers R, Joosten HJ, Soetaert W, Desmet T (2011). Increasing the thermo stability of sucrose phosphorylase by a combination of sequence-and structure-based mutagenesis. Protein Eng. Des. Sel. 24(11):829-834 |
|
|
Heidrich K, Otto A, Behlke J, Rush J, Wenzel KW, Kriegel T (1997). Autophosphorylation-inactivation site of hexokinase 2 in Saccharomyces cerevisiae. Biochem. 36(8):1960-1964 |
|
|
Jochens H, Aerts D, Bornscheuer UT (2010). Thermostabilization of an esterase by alignment-guided focussed directed evolution. Protein Eng. Des. Sel. 23(12):903-909. |
|
|
Joosten HJ (2007). 3DM: from data to medicine. PhD Thesis, University of Wageningen, Wageningen, The Netherlands. |
|
|
Krieger E, Koraimann G, Vriend G (2002). Increasing the precision of comparative models with Yasara NOVA-a self-parameterizing force field. Proteins: Struct. Funct. Genet. 47:393-402. |
|
|
Kuipers R, van den Bergh T, Joosten H-J, Lekanne dit Deprez RH, Mannens MM, Schaap PJ (2010a). Novel tools for extraction and validation of disease-related mutations applied to fabry disease. Hum. Mutat. 31(9):1026-1032, |
|
|
Kuipers RK, Joosten HÄ, van Berkel WJ, Leferink NG, Rooijen E, Ittmann E, van Zimmeren F, Jochens H, Bornscheuer U, Vriend G (2010b). 3DM: systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins: Structure, Function, and Bioinformatics 78(9):2101-2113. |
|
|
Kuser PR, Krauchenco S, Antunes OAC, Polikarpov I (2000). The High Resolution Crystal Structure of Yeast Hexokinase PII with the Correct Primary Sequence Provides New Insights into Its Mechanism of Action. J. Biol. Chem. 275(27):20814-20821. |
|
|
Moreno F, Herrero P (2002). The hexokinase 2-dependent glucose signal transduction pathway of Saccharomyces cerevisiae. FEMS Microbiol. Rev. 26(1):83-90 |
|
|
Nishimasu H, Fushinobu S, Shoun H, Wakagi T (2007). Crystal Structures of an ATP-dependent Hexokinase with Broad Substrate Specificity from the Hyperthermophilic Archaeon Sulfolobus tokodaii. J. Biol. Chem. 282(13):9923-9931. |
|
|
Nobili A, Gall MG, Pavlidis IV, Thompson ML, Schmidt M, Bornscheuer UT (2013). Use of 'a small but smart' libraries to enhance the enantioselectivity of an esterase from Bacillus stearothermophilus towards tetrahydrofuran-3-yl acetate. FEBS J. 280(13):3084-3093. |
|
|
Trott O, Olson AJ (2010). Auto Dock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31(2):455-461. |
|
|
Wang M, Si T, Zhao H (2012). Biocatalyst development by directed evolution. Bioresour. Technol. 115:117-125. |
|
|
Wijma HJ, Janssen DB (2013). Computational design gains momentum in enzyme catalysis engineering. FEBS J. 280(13):2948-2960. |
|
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


