ABSTRACT
Despite the widely spread acceptance that the enthalpy of DNA duplex unfolding does not depend on temperature and is greater for the CG base pair held by three hydrogen bonds than for the AT base pair held by only two hydrogen bonds, direct calorimetric measurement has shown that the enthalpic and entropic contributions of both base pairs are temperature dependent and at all temperatures they are greater for the AT base pair. The larger enthalpic and entropic contributions of the AT base pair result from water that is fixed by this pair in the minor groove of DNA and is released at duplex dissociation, while their dependence on temperature results from hydration of the apolar surfaces of bases that become exposed upon duplex dissociation. Analysis of the obtained thermodynamic characteristics of unfolding/refolding of the DNA duplexes of various compositions shows that the enthalpic contribution of base pairing is negligibly small, while the entropic is considerable. Thus, the DNA base pairing is the entropy driven process but it is conjugate with the bases stacking that is the enthalpy driven. The last is responsible for about 50% of duplex stabilizing Gibbs energy and for almost 100% of its enthalpy, that is for the total heat of DNA melting.
Key words: DNA, stability, hydrogen bonding, base stacking, hydration.
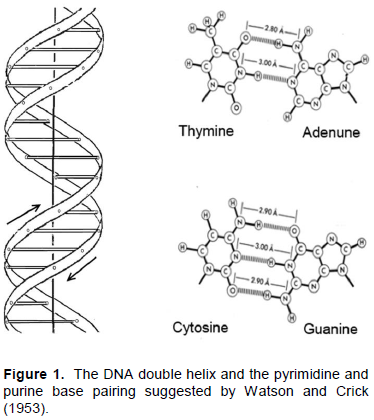
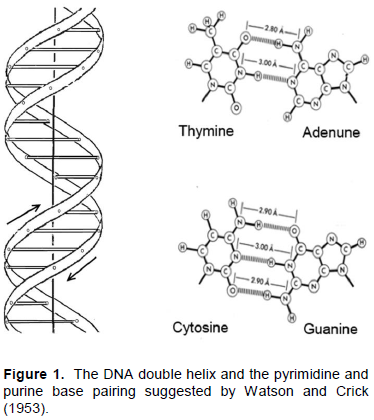
The appearance of the Watson and Crick model of DNA in 1953 was the greatest event in biological science as it explained the mechanism of coding genetic information and its multiplication (Figure 1). Moreover, it has suggested the physical ground for the DNA double helix that appeared to be supported by the hydrogen bonds between the conjugate base pairs, two between the A and T and three between the C and G bases. This seems to be confirmed by the optical observation that increase of the CG content leads to increase of the DNA duplex thermostability (Marmur and Doty, 1962). Since then there were many attempts to estimate the thermodynamic contribution of base pairing to the double helix maintenance measuring the heats of melting of the synthetic polynucleotides by the conventional calorimeters for liquids. Although steering the highly viscose solutions of polynucleotides in these instruments was a problem, they seemed to confirm expectation that enthalpic contribution of the C and G base pairing exceeds that of the A and T, as was believed assuming that the double helix is maintained only by the hydrogen bonds between these two bases pairs (Krakauer and Sturtevant, 1968; Neuman and Ackerman 1969; Breslauer and Sturtevant, 1977; Breslauer et al., 1986; Chalikian et al., 1999). It was suggested also that certain contributions to the DNA duplex formation might be expected from compact packing of the flat bases (Breslauer et al., 1986; Sugimoto et al., 1996; SantaLucia, 1998; Yakovchuk et al., 2006). Doubts in the experimental ground for all these suggestions stimulated appearance of more precise calorimetric instruments designed for studying the viscous and highly deficient solutions: the Nano-DSC and Nano-ITC (Privalov, 2012). These instruments permitted to start detailed thermodynamic investigation of the short synthetic DNA duplexes of certain compositions (Vaitiekunas et al., 2015).
CALORIMETRY OF THE DNA DUPLEXES
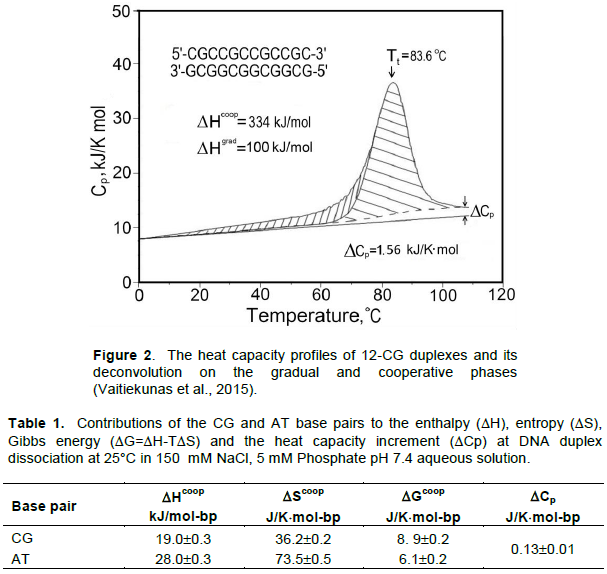
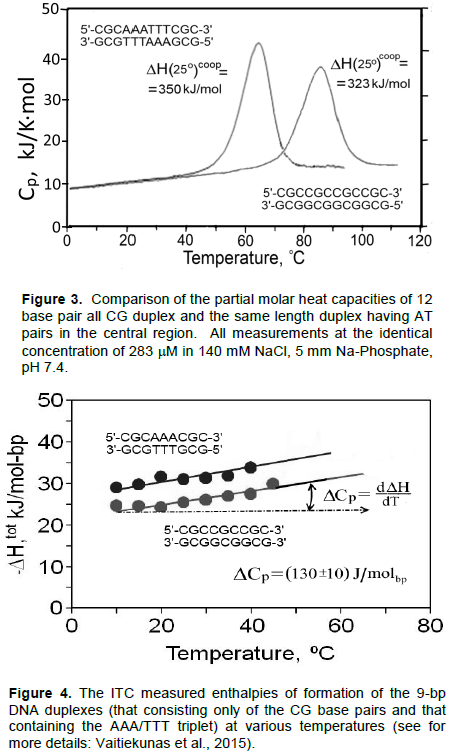
Using the Nano-DSC (Differential Scanning Calorimeter) it was found that the temperature inducing unfolding/refolding of the DNA duplex is not a simple process as it was supposed to be (Jelesarove et al., 1999; Privalov, 2012). It consists of gradually increasing torsion fluctuations of the duplex followed by a phase of cooperative dissociation of forming it strands proceeded with large heat absorption and resulting in significant heat capacity increment (Figure 2). Thus, since the heat capacity is a temperature derivative of enthalpy, the enthalpy of duplex dissociation appeared to be temperature dependent. Surprisingly this heat capacity increment was found to be identical for both the AT and CG base pairs, amounting to (0.13±0.01)×kJ/K×mol-bp (Table 1). Furthermore, although duplexes containing AT base pairs melt, as expected, at lower temperatures than those consisting only of CG pairs, absolutely unexpected had less stable duplexes of the same length but containing AT pairs melt with a higher heat effect (Figure 3). Thus, contrary to the expectation, the enthalpic contribution of the AT base pair exceeds that of the CG base pair. Since the AT-containing duplex melts at a lower temperature than the same length duplex but entirely of CG pairs, one can conclude that the entropic contribution of the AT base pair also exceeds significantly that of the CG base pair.
The above results obtained by Nano-DSC were confirmed by the titration experiments carried with Nano-ITC (Isothermal Titration Calorimeter). It appeared that the heat effect of association of the strands of identical length but differing in the presence of AT base pairs exceeds that of the purely CG duplex at all studied temperatures (Figure 4).
It is notable that the enthalpies of formation of both duplexes are temperature dependent: they increase with temperature rise with a similar slope, ¶DH/¶T=(0.13±0.01) kJ/K×mol-bp, that is with exactly the same heat capacity increment, DCp, as was found analyzing the melting profiles of the duplexes. Comparison of the enthalpic and entropic contributions of the base pairs is less straightforward because of the near-neighbors effect that occurs at the border of the AT and CG base pairs (Borere et al., 1974); however, using the long enough non-interrupted AT sequences these values can be determined with reasonable accuracy. Thus, it followed from the ITC and DSC data that extrapolated to standard temperature 25°C the enthalpic and entropic contributions of the CG and AT base pairs are:
DHCG(25°C)=(19.0±0.2)×kJ/mol-bp, and
DHAT(25°C)=(28.0±0.3)×kJ/mol-bp (1)
DSCG(25°C)=(36.2±0.2)×J/K×mol-bp, and
DSAT(25°C)=(73.5±0.5)×J/K×mol-bp. (2)
It appears therefore that the duplex stabilizing effect of the CG base pair is larger than that of the AT not because its enthalpic contribution is larger but because its entropic contribution is smaller (Privalov and Crane-Robinson, 2018a, b). The question is: why is the entropic and also enthalpic contribution of the AT base pair larger than that of the CG?
ROLE OF WATER
As follows from (1) and (2), the difference between the CG and AT base pairs contributions amounts to 6.0 kJ/mol-bp in enthalpy and 28.6 J/K×mol-bp in entropy. Surprisingly these values are very close to the enthalpy and entropy of water freezing which are 6.0 kJ/mol and 22.0 J/K×mol. The close correspondence between the found differences in the base pairs contributions to the duplex stability and the ice melting characteristics suggest that the difference between the AT and CG base pairs thermodynamic contributions is caused by the water molecule (Privalov and Crane-Robinson, 2018a, b).
Water specifically fixed by the AT base pairs in the minor groove of DNA was indeed noticed crystallographically (Drew and Dickerson, 1981). It was found that these water molecules are fixed by the N3 of A and O2 of T bases of the AT pair (Kopka et al., 1983; Prive et al., 1987; Mandal et al., 2016). Release of the tightly bound in the minor groove water into the bulk solution should result in positive contributions to both the enthalpy and entropy of DNA melting. Excluding this water contribution from the calorimetrically measured enthalpy and entropy of the A and T bases, the net enthalpic contributions of these bases appear then to be almost similar to that of C and G:
DHCG(25°C) = (19.0±0.2)×kJ/mol-bp and DSCG(25°C) = (36.2±0.2)×J/K×mol-bp (3)
DHAT(25°C) = (22.0±0.3)×kJ/mol-bp and DSAT(25°C) = (51.0±0.2)×J/ K×mol-bp (4)
FORCES HOLDING THE DNA BASE PAIRS
Assuming, as usually believed, that the DNA double helix is maintained by the hydrogen bonds between the conjugate bases and dividing the obtained enthalpy, entropy and the Gibbs energy values of the CG base pair (Table 1) on the number of hydrogen bonds between these bases, one finds as if a single hydrogen bond contributes about 6.3 kJ/mole-bp in enthalpy, 12 kJ/K×mole-bp in entropy and 3 kJ/mol-bp in Gibbs energy. These values largely exceed the expected for the brake of a single hydrogen bond between the polar groups in the aqueous media: after Pauling (1960) the enthalpy of a single hydrogen bond between the polar groups of protein in aqueous solution is expected to be slightly positive, while the entropy be negative since the exposed polar groups are increasing order in the surrounding water. Comparison of the given in (3) and (4) enthalpies of the CG base pair and the corrected on water AT base pair shows indeed that they do not differ much. Since the CG base pair is held by three hydrogen bonds, while the AT by two, one can conclude that the enthalpic component of the base pair hydrogen bonding is indeed negligibly small. Comparison of the entropic contribution shows that it is negative and amounts to -(50±0.3)J/K×mol per bond. Therefore, at standard temperature 25oC=298.16 K, a single hydrogen bond provides about DG=-DS´T=(5.0±0.3)J/Kmol-bp´298.16K =(1.6±0.1) kJ/mol to the Gibbs energy of base pair, which is just what is usually expected for the hydrogen bonds maintaining proteins structure in aqueous solution. It appears then that the Gibbs energy of a single CG base pair (which is held by 3 hydrogen bonds) amounts to 4.8 kJ/mol, while of a single AT base pair (which is held by two hydrogen bonds) it amounts to 3.2 kJ/mol. It is notable, however, that these two base pairs provide almost nothing to the enthalpy of duplex stabilization. This immediately raises the question: what is then the source of the calorimetrically observed large heat effect of DNA duplex melting, that is the source of the enthalpy of DNA duplex dissociation?
THE ENTHALPY OF DNA UNFOLDING
The calorimetrically observed large enthalpy of DNA melting might result (a) from release of the water fixed in the minor groove of DNA; (b) from unpacking the bases in the DNA duplex. The first might provide about 6.0 kJ per mole of AT base pair but the rest of the DNA enthalpy might result only from melting the stacked bases. One could expect that unpacking the bases stack might be responsible not only for the large enthalpy of DNA melting but also for its clear dependence on temperature, that is for the heat capacity increment specific for the DNA melting (Figure 2 and Table 1).
The experimentally observed significant heat capacity increment at DNA duplex dissociation presents striking controversy in the DNA energetics. It is known that hydration of the polar groups results in their partial heat capacity decrement, in contrast to the apolar group’s hydration that results in the partial heat capacity increment (Kauzmann, 1959; Privalov and Gill, 1988; Privalov and Makhatadze; 1992; Spolar et al., 1992; Makhatadze and Privalov, 1995). According to Privalov et al. (1992) the heat capacity effect of hydration of the apolar and polar groups is expressed by the equation:

Thus, break of the hydrogen bindings between the coupled bases of DNA, which results in appearance of the new charged groups, should proceed with the DNA partial heat capacity decrement. In fact, as demonstrates Figure 2, at the DNA unfolding-dissociation its partial heat capacity increases. The clear heat capacity increment at the DNA duplex dissociation shows that this process proceeds not only with break of the hydrogen bonds between the polar groups and exposure of these groups to water but also with breaking the contacts between the apolar groups tightly packed in the DNA interior and exposure of these apolar groups to water.
SURFACES EXPOSED UPON DNA DUPLEX DISSOCIATION
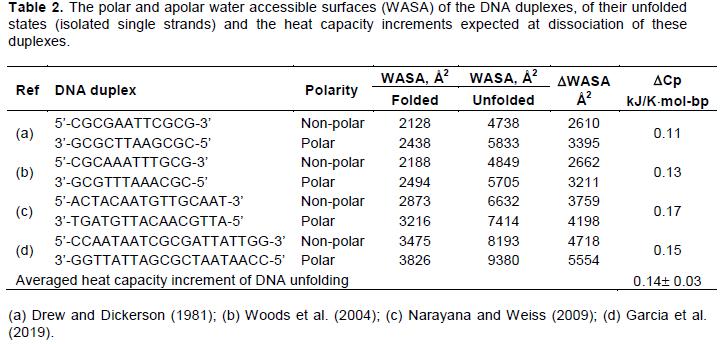
At the present time there are several well defined crystallographic structures of DNA duplexes that permit estimation of their exposed surfaces. Unfortunately, we do not have the structure of the unfolded DNA since its separated strands, being highly flexible, are in extensive thermal motion, fluctuate. Nevertheless, approximating unfolded DNA with its isolated single strands, one might determine by the Nacess program the approximate water accessible surface areas (WASA) of the polar and non-polar groups (Dragan et al., 2019). Such analysis carried with the several DNA duplexes showed that their unfolding results indeed in significant increase of the exposed apolar and polar surfaces (Table 2). Increase of the polar surfaces upon DNA unfolding might result mainly at disruption of the hydrogen bodings between the paired polar bases, while increase of the apolar surfaces might result only from dissociation of the tightly stacked bases resulting in exposure of their apolar flat surfaces to water. It is remarkable that in the unfolded DNA the exposed polar and apolar surfaces are almost similar in area. However, since the intrinsic heat capacity effect of apolar surface hydration largely exceeds that of the polar groups hydration (Equation 5), the overall heat capacity effect of DNA duplex unfolding is positive: the averaged expected heat capacity effect of both the AT and CG base pairs dissociation appears to be DCp=(0.14±0.03)×kJ/K×mol-bp. This value is surprisingly close to the calorimetrically measured heat capacity increment at DNA duplex unfolding, (0.13±0.01) kJ/K×mol-bp. Close correspondence of the measured and calculated heat capacity effects show that the used approximation of the unfolded DNA duplex by its isolated single strands is valid. Moreover, this correspondence shows that upon heating DNA not only the polar groups involved in hydrogen bonding of the conjugate bases but also the apolar surfaces of those bases become exposed to water.
It should be noted that the recalculated per gram the specific heat capacity increment at DNA duplex unfolding is significantly smaller than the specific heat capacity increment at globular proteins unfolding (Makhatadze and Privalov, 1995; Privalov, 2012). This shows that the concentration of the apolar groups in the DNA interior is significantly lower than in the globular proteins. The high concentration of apolar groups in proteins is just what makes them globular. It appears, however, that the contacts between the apolar bases in the DNA double helix are also playing essential role in its construction.
CONTRIBUTION OF THE BASES TO THE DNA DUPLEX STABILITY
Earlier studies of the DNA duplex stability, affected by the Watson and Crick model of DNA and by the first optical observations that the presence of three hydrogen bonded CG base pair increases the DNA duplex stability encouraged the belief that H-bonding is the primary determinant of duplex stability and thus of its thermodynamics. However, the hydrogen bonds, which are entropic, cannot be responsible for the large enthalpy of DNA unfolding and more so for its dependence on temperature, that is the heat capacity increment specific for the DNA melting. It appears, therefore, that there should be in the DNA duplex some other source of the enthalpy except the hydrogen bonding between the conjugate bases. As such might be only the tightly packed bases in the DNA duplex. The flat apolar surfaces of these bases suggest extended van der Waals contacts between them. Disruption of these contacts should require considerable enthalpy, while exposure of their apolar surfaces into water should result in the significant heat capacity increment. It is just this heat effect and the heat capacity increment that are calorimetrically recorded upon DNA melting (Figure 2). The question is then: how much Gibbs energy is provided by the bases stacking to the DNA duplex stabilization?
As shown in Section 4, the Gibbs energy of CG base pair, which is held by three hydrogen bonds, amounts to (4.8±0.3) kJ/mol-bp, while the overall Gibbs energy of this base pair is (8.9±0.2) J/K×mol-bp (Table 1). The difference between these two values, about 4.1 kJ/mol-bp, might be provided only by the stacked bases. Thus, it appears that the base stacking is responsible for about 46% of the overall Gibbs energy of the CG base pair. Similarly, the two hydrogen bonds might provide only 3.2 kJ/mol-bp to the Gibbs energy of the AT base pair, while the overall Gibbs energy of this base pair is (6.1±0.2) 1J/K×mol-bp (Table 1). It appears, therefore, that in the case of AT base pair the base stacking is responsible also for almost 50% of the whole Gibbs energy of AT base pair. It is remarkable, however, that in both cases stacking of bases is responsible for almost all enthalpy of DNA melting.
It appears that the base pairing and the base stacking are two tightly interconnected processes. Indeed, pairing the bases requires their proper orientation; but proper orientation of the bases leads to their stacking; and vice versa: the base stacking assumes proper orientation of the conjugate bases that is required for their hydrogen bonding. Thus, these two steps represent a single cooperative act of the DNA double helix formation. Cooperation of the base pairing and base stacking explains the extreme efficiency of the DNA double helix propagation that proceeds with raise of its rigidity. In this cooperative folding process, while the hydrogen bonds between the conjugate bases are of critical importance for the proper adjustment of the two complementary strands of DNA, the formed double helix is reinforced by the simultaneous stacking of the apolar bases.
The authors have not declared any conflict of interests.
The author thank Paulius Vaitiecunas (TA Instruments), Colyn Crane-Robinson (University of Portsmouth, UK), Ilian Jelesarove (University of Zurich, Switzerland) and Anatoly Dragan (University of Kiev, Ukraine) for participation in the calorimetric studies of the DNA duplexes at Johns Hopkins University.
REFERENCES
|
Borere PN, Dengler B, Tinoco Jr-I, Uhlenbeck OC (1974). Stability of ribonucleic acid double-stranded helices. Journal of Molecular Biology 86:843-853.
Crossref
|
|
|
|
Breslauer KJ, Sturtevant JM (1977). A calorimetric investigation of singlestranded base stacking in ribo0nucleotide A7. Biophysical Chemistry 7:205-209.
Crossref
|
|
|
|
|
Breslauer KJ, Frank R, Blocker H, Marky LA (1986). Predicting DNA duplex stability from the base sequence. Proceedings of the National Academy of Sciences of the United States of America 83:3746-3750.
Crossref
|
|
|
|
|
Chalikian TV, Volker J, Plum GE, Breslauer KJ (1999). A more unified picture for the thermodynamics of nucleic acid duplex melting: a characterization by calorimetric and volumetric techniques. Proceedings of the National Academy of Sciences of the United States of America 96:7853-8758.
Crossref
|
|
|
|
|
Dragan AI, Crane-Robinson C, Privalov PL (2019). Thermodynamic of DNA: heat capacity changes on duplex unfolding. European Biophysics Journal. in press.
Crossref
|
|
|
|
|
Drew HR, Dickerson RE (1981). Structure of a B-DNA dodecamer. III. Geometry of hydration. Journal of Molecular Biology 151:535-556.
hCrossref
|
|
|
|
|
Garcia S, Acosta-Reyes FJ, Saperas N, Campos JL (2019). Crystal Structure of AT -rich DNA 20mer. To be published.
|
|
|
|
|
Jelesarove I, Crane-Robinson C, Privalov PL (1999). The energetics of HMG box interuction with DNA. Thermodynamic description of teh target DNA duplexes. Journal of Molecular Biology 294:981-995.
Crossref
|
|
|
|
|
Kauzmann W (1959). Some factors in the interpretation of protein denaturation. Advances in Protein Chemistry 14:1-63.
Crossref
|
|
|
|
|
Kopka ML, Fratini AV, Drew HR, Dickerson RE (1983). Ordered water structure around a B-DNA dodecamer. Journal of Molecular Biology 163:129-146.
Crossref
|
|
|
|
|
Krakauer H, Sturtevant JM (1968). Heats of the helix-coil transitions of the polyA-polyU complexes. Biopolymers 6:491-512.
Crossref
|
|
|
|
|
Mandal PK, Collie GW, Srivastava SC, Kauffmann B, Huc I (2016). Structure elucidation of the Pribnow box consensus promoter sequence by racemic DNA crystallography. Nucleic Acids Res. 44:5936-5943.
Crossref
|
|
|
|
|
Makhatadze GI, Privalov PL (1995). Energetics of protein structure. Advances in Protein Chemistry 47:307-425.
Crossref
|
|
|
|
|
Marmur J, Doty P (1962). Determination of the base composition of deoxyribonucleic acid from its thermal melting temperature. Journal of Molecular Biology 5:109-118.
Crossref
|
|
|
|
|
Narayana N, Weiss MA (2009). Crystallographic Analysis of a Sex-Specific Enhancer Element: Sequence-Dependent DNA Structure, Hydration, and Dynamics. Journal of Molecular Biology 385:469-490.
Crossref
|
|
|
|
|
Neuman E, Ackerman T (1969). Thermodynamic investigation of the Helix-Coil transition of a polyribonucleotide system. Journal of Physical Chemistry 73:2170-2178.
Crossref
|
|
|
|
|
Pauling L (1960). The nature of the chemical bond. Cornell University Press.
|
|
|
|
|
Privalov PL (2012). Microcalorimetry of macromolecules. The physical bases of biological structures. Wiley & Sons.
Crossref
|
|
|
|
|
Privalov PL, Crane-Robinson C (2018a). Translational entropy and DNA duplex stability. Biophysical Journal 114:15-20.
Crossref
|
|
|
|
|
Privalov PL, Crane-Robinson C (2018b). Forces maintaining the DNA double helix and its complexes with transcription factors. Progress in Biophysics and Molecular Biology 135:30-48.
Crossref
|
|
|
|
|
Privalov PL, Gill SJ (1988). Stability of protein structure and hydrophobic interaction. Advances in Protein Chemistry 39:191-234.
Crossref
|
|
|
|
|
Privalov PL, Makhatadze GI (1992). Contribution of hydration and non-covalent interactions to the heat capacity effect on protein unfolding. Journal of Molecular Biology 224:715-723.
Crossref
|
|
|
|
|
Prive GG, Heinemann U, Chandrasegaran S, Kan LS, Kopka ML, Dickerson RE (1987). Helix geometry, hydration, and G.A mismatch in a B-DNA decamer. Science 238:498-504
Crossref
|
|
|
|
|
SantaLucia J-Jr (1998). A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proceedings of the National Academy of Sciences of the United States of America 95:1460-1465.
Crossref
|
|
|
|
|
Spolar RS, Livingston JR, Record MT (1992). Contributions to Thermodynamic Functions of Protein Folding from the Removal of Nonpolar and Polar Surface from Water. Biochemistry 31:3947-3955
Crossref
|
|
|
|
|
Sugimoto N, Nakano S, Yoneyama M, Honda K (1996). Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Research 24:4501-4505.
Crossref
|
|
|
|
|
Vaitiekunas P, Crane-Robinson C, Privalov PL (2015). The energetic basis of the DNA double helix: a combined microcalorimetric approach. Nucleic Acids Research 43:8577-8589.
Crossref
|
|
|
|
|
Watson JD, Crick FHC (1953). A structure for deoxyribonucleic acid. Nature 171:737-738.
Crossref
|
|
|
|
|
Woods KK, Maehigashi T, Howerton SB, Sines CC, Tannenbaum S, Williams LD (2004). High-resolution structure of an extended A-tract. Journal of the American Chemical Society 126:15330-15331.
Crossref
|
|
|
|
|
Yakovchuk P, Protozanova E, Frank-Kamenetskii MD (2006). Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Research 34:564-574.
Crossref
|
|