The present study was conducted to assess the molecular discrimination of field isolates of Trypanosoma congolense and Trypanosoma vivax of Nigeria origin in vitro (PCR) whether there will be genetic alteration of the parasites as the infections of the isolates in Yankasa sheep progresses from acute through relapses to chronic stages. A total of thirty Yankasa sheep were acquired from Kastina State in the northern part of Nigeria, screened for haemo-, ecto- and endo-parasites, treated against anaplasmosis, coccidiosis and conditioned in arthropod-free pens for two weeks. They were randomly divided into five groups of six animals each. Two groups each were infected intravenously with approximately 2.0×106 of T. vivax and T. congolense. One group served as uninfected control. A group each from T. vivax-infected and T. congolense-infected groups were treated with trypanocide 6 days post parasitaemia. The other infected groups were left untreated. The animals were monitored for 8 weeks post infection (pi). Incubation period of 6 days was recorded for both parasite species. Multiplex primers polymerase chain reaction (PCR) and T. vivax species-specific PCR confirmed that the isolates used were T. congolense and T. vivax at molecular band sizes of 750 and 175 bp, respectively. Randomly amplified polymorphic DNA (RAPD)-PCR showed that the band sizes associated with the chronic form of the infection of T. congolense differed from those of the acute and relapse forms, whereas the band weights of the relapse form of the T. vivax differed from those of acute and chronic forms. Molecular characterization of T. vivax and T. congolense revealed differences through the variations in band weights of the parasites derivatives in the acute, chronic and relapse infections and this may translate into differences in antigenicity.
African trypanosomosis is currently resurgent across a great part of tropical Africa, reaching epidemic levels in many places (Stich et al., 2002; Delespaux et al., 2008; Ogbaje et al., 2015). Trypanosomavivax and Trypanosoma congolense are the major species responsible for African Animal Trypanosomosis (AAT) or Nagana in Nigeria and sub-Saharan African (Takeet et al., 2013; Isaac et al., 2016; Morrison, 2016). T. vivax and T. congolense infect large variety of domestic and wild animals (D’avila et al., 1997; Auty et al., 2012).
Polymerase chain reaction (PCR) was introduced for detection of trypanosome DNA in biological samples. PCR have been proved to be highly sensitive and specific and has been widely used in the detection of trypanosomes (Masiga et al., 1992; Ventura et al., 2001). The techniques have been found to be specific and sensitive for large scale analysis of trypanosome samples (Hide and Tait, 1991). It is also capable of detecting minute quantities of DNA of specific pathogens through amplification of a defined DNA segment and the discrimination in one reaction between different organisms even if they are closely related (Hatta and Smits, 2007).
Previous studies showed that the sensitivity of PCR in detecting trypanosomes is about two times higher than that of Ag-ELISA and four times higher than with the parasitological techniques, especially the Wet blood film and thin blood smear (Narendra et al., 2004; Clausen et al., 1998; Solano et al., 1999).
The accurate identification of a parasite to the species level has major implications for various aspects in veterinary parasitology, including diagnosis as well as treatment and control. The resurgence and difficulty in the control of the disease due to antigenic variation and development of drug-resistant strains have necessitated the need for more research in the study of the gene and nature of DNA elements within the genome of the parasites for possible vaccine and new drug development (Greif et al., 2013; Jackson et al., 2015; Morrison, 2016).
The aim of this research was to carry out molecular characterization of T. congolense and T. vivax isolates of Nigeria origin and to check whether there will be genetic alteration of the parasites as the infections of the isolates in Yankasa Sheep progresses from acute through relapses to chronic stages.
Parasite isolates used
The isolates of T. vivax and T. congolense used in this study were isolated from sedentary white Fulani cattle in Makurdi, Benue State and Idon, Southern part of Kaduna State, Nigeria, respectively. About sixteen cattle were selected from the herd in Makurdi and twenty one at Idon based on their history of dullness, pica appetites by the herd men and high pyrexia on physical examinations.
Experimental animals
Thirty (30) Yankasa sheep, aged between 2 and 3 years were used for the study. The sheep were purchased from an open market at Karfur in Kastina State considered to be free of tsetse flies and consequently pathogenic Trypanosoma species. On arrival, the animals were screened using microscopy for endoparasites, haemoparasites and also through physical examination for presence of ecto-parasites. Two milliliters (2 ml) of blood was obtained from the jugular vein of each of the sheep, and examined for haemoparasites using wet mount, thin blood smear and microhaematocrit methods as described by Woo (1971). Three grams of faeces was scooped from the rectum of each sheep using a clean polythene bag and taken to Helminthology Laboratory, processed and examined for helminthes eggs using floatation and sedimentation methods as described by Cole (1986).
All the experimental sheep were dewormed orally using Albendazole® at the dose rate of 7.5 mg/kg. Ecto-parasites infestations were treated and controlled with deltamethrin® pour-on preparation and asuntol® spray. Those found with Anaplasma infections were treated with oxytetracycline long acting at the dose rate of 20 mg/kg body weight. Amprolium was administered to the ones that were infected with Coccidia for 5 days according to the manufacturer’s recommendation. The sheep were also vaccinated each with 1 ml subcutaneous injection of monoclonal pestes de petit ruminantes (PPR) vaccine, against PPR. The sheep were kept in arthropod-free pens of the Department of Veterinary Parasitology and Entomology, Faculty of Veterinary Medicine, Ahmadu Bello University, Zaria and pre-conditioned for two weeks before the commencement of the experiment.
The experimental animals were tagged and randomly divided into five groups (A, B, C, D and E) of six animals each per group. Base line data were obtained from each of the animal in all the groups for a period of one week prior to infections. Two different sheep were used as donor animals, one sheep each for T. vivax and T. congolense to multiple the parasites. Each of the sheep in groups A and B was inoculated through the jugular vein with 2 ml of blood containing approximately 2.0×106 T. vivax as quantified using the improved Naubauer haemocytometer (Petana, 1963). Each animal in groups D and E were also inoculated through the jugular vein with 2 ml of blood containing approximately 2.0×106 T. congolense as quantified using the improved Naubauer haemocytometer. Group C served as uninfected control. Groups A and E animals were treated with diminazene aceturate at the height of parasitaemia (++++).
The DNA profiles of the parasites were examined at different stages of the infections (acute, relapse and chronic) using randomly amplified polymorphic DNA analysis (RAPD-PCR). The following samples/forms of the parasites were collected from only trypanocidal treated animals and preserved at -20°C and later subjected to Trypanosoma Genus specific PCR, Trypanosoma species-specific PCR and RAPD-PCR analysis:
1. Acute form of the T. vivax and T. congolense (a week post infection)
2. Relapse forms of the T. vivax and the T. congolense (first phase of parasitaemia after treatment)
3. Chronic form of the infections with T. vivax and T. congolense (8 week post infection)
The Trypanosoma Genus specific PCR primers (Table 1) for Trypanosoma and T. vivax species-specific primers were supplied by Inqaba Biotechnical Industries LTD, Hatfield- Pretoria, S/Africa and RAPD-PCR primers were obtained from Bioneer-Biotech Company, Washington DC, USA. The experiment was divided into three different phases:
1. Each of the two isolates was subjected to Trypanosoma Genus specific PCR using primers for identification of different species and to rule out possibility of mixed infections in any of the samples.
2. T. vivax species-specific PCR was conducted on the two field isolates for T. vivax species identification of the fields samples (confirmatory test for T. vivax isolate).
3. RAPD-PCR was conducted on the three different forms (the acute, relapse and chronic forms) of the two isolates.
Identification of different Trypanosoma species using Trypanosoma genus specific primers PCR
DNA extraction from whole blood: DNA was extracted using QIAamp® DNA Mini Kit (Qiagen, Germany). The method was conducted as described by Desquesnes et al. (2001). Briefly, a pair of primers designed for ribosomal DNA was used. The amplification was performed in a final volume of 50 μl containing 5 μl of isolated DNA (template), (20 µM = 1 µL) of each primer (Desquesnes et al., 2001), 2 mM of each dNTP, 32.5 µL of Nuclease-free water and 0.5 unit of Taq DNA polymerase. Initial denaturing step at 94°C for 5 min was followed by 30 amplification cycles. Each cycle consisted of a denaturation step at 94°C for 30 s, an annealing step at 58°C for 30 s, 54°C for 30 s and an extension step at 72°C for 1 min, and a final extension at 72°C for 10 min. The total number of cycles was 42 (4 cycles for 58°C, 8 cycles for 56°C and 30 cycles for 54°C).
The amplified products of the PCR were resolved on 2% agarose gel at 100 V for 60 min containing 1.0 mg/μl of ethidium bromide. The gels were observed on ultraviolet light and photographed.
Trypanosoma vivax species-specific PCR
In order to confirm the presence or absence of T. vivax in any of the samples, because of the low sensitivity of the Trypanosoma Genus specific primers PCR, they were subjected to T. vivax- specific primers PCR.
DNA extraction from whole blood: The DNA was extracted using QIAamp® DNA Mini Kit (Qiagen, Germany).
Primers: A pair of specific primers for T. vivax was used in this study as described by Desquesnes et al. (2001)
DNA amplification
The amplification was performed in a final volume of 25 μl containing 2 μl of isolated DNA (template), (1 µM = 1 µL) of the primer (TVW A- dGTG CTC CAT GTG CCA CGT TG TVW B- dCAT ATG GTC TGG GAG CGG GT), 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 200 µM of dNTPs, 10 µL of Nuclease free water, chelex 10 µl. Initial denaturing step was carried out at 94°C for 5 min and then 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 60 s and extension at 72°C for another 5 min.
RAPD-PCR of T. vivax and T. congolense
Stabilates of the two isolates (T. vivax and T. congolense) were retrieved from the freezer (-20°C).
DNA extraction from the T. vivax and T. congolense isolates
Two hundred and fifty microlitres (250 µl) of blood was mixed with 250 µl of lysis buffer (0.31 M sucrose, 0.01 M Tris-HCl, pH 7.5, 5 mM MgCl2, 1% Triton X-100). The mixture was centrifuged at 15,000 ×g for 20 s, the supernatant was removed and 500 µl of lysis buffer was added and mixed by vortexing. Centrifugation and addition of lysis buffer was repeated twice. After the last centrifugation and removal of the supernatant, the pellet was re-suspended in 250 µl of 1× PCR buffer (10 mM Tris-HCl, pH 8.4, 50 mM KCl, 1% Triton X-100) and 1.5 µl of proteinase-K (10 mg/ml) was added, mixed and incubated at 56°C for 1 h. Finally, the mixture was incubated at 95°C for 10 min to inactivate the proteinase-K. The resulting eluate was stored frozen at -20°C until used (Clausen et al., 1998).
Characterization of the extracted DNA using randomly amplified polymorphic DNA PCR (RAPD-PCR)
The extracted DNAs from the two isolates were subjected to RAPD-PCR using short random primers as described by Welsh and McClelland (1990) and modified by Tibayrenc et al. (1993). Genomic DNA samples (20 ng) were amplified in 60 µl of specific buffer (10 mM Tris- HCl, 50 mM KCl, 1.5 mM MgCl2, pH 8.3), in the presence of 0.2 µM of primers, 4x 100 mM, dNTP and 0.9 µl of Taq DNA polymerase. The RAPD-PCR was based on the use of twenty base pair primers (Tibayrenc et al., 1993). The total number of amplification was 45 cycles at 94°C for 1 min and 60°C for 2 min and a final elongation at 72°C for 7 min. The amplification was repeated three times for the pair of the primers. The amplified product was separated by electrophoresis on 1.6% agarose gels with TAE buffer (40 mM Tris- acetate pH 7.5, 1 mM EDTA) at 5 V/cm and DNA fragments were visualized after staining with ethidium bromide. The patterns of amplified fragments of each isolate were used to compare the isolates.
Identification of different species of trypanosomes (mixed infections) using multiplex Trypanosoma primers-PCR
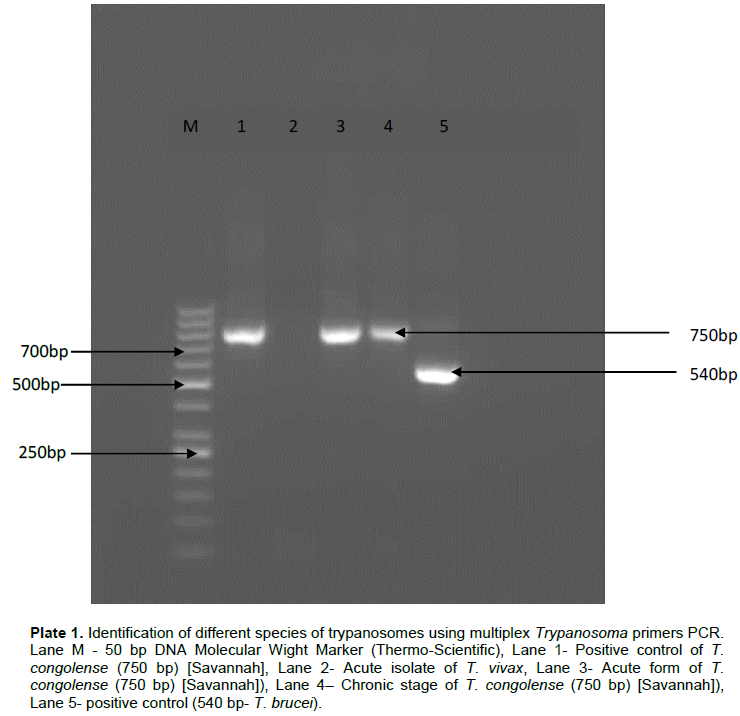
The result shows that the isolate from the Northern Guinea savannah (Idon in Kaduna State) is purely Savannah type of T. congolense with a band of 750 bp (Plate 1). Sample from Southern Guinea Savannah (Makurdi-Benue state) did not contain T. congolense.
T. vivax species-specific PCR
In order to confirm the presence or absence of T. vivax in any of the samples (Kaduna and Makurdi), the samples were subjected to T. vivax-specific primers PCR. The acute and relapse samples of T. vivax (Makurdi sample) yielded the species specific amplicon of 175 bp (Plate 2), indicating pure T. vivax.
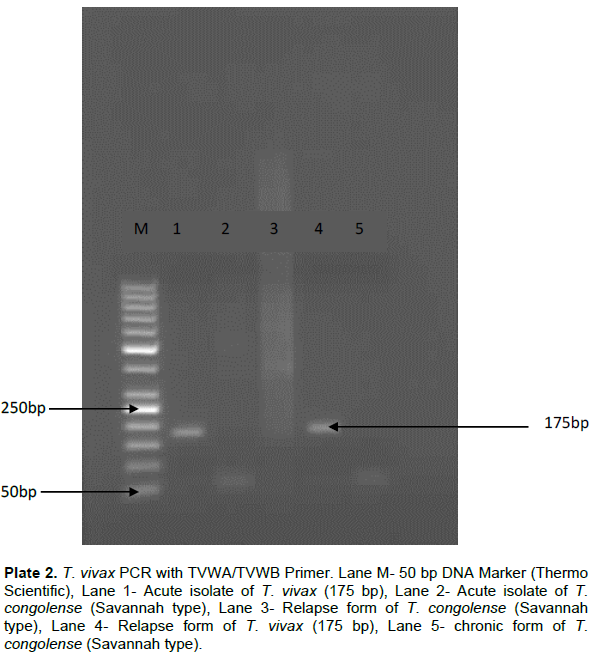
The results revealed that sample from Southern Guinea Savannah (Makurdi) was pure T. vivax isolates since it was negative to multiplex primers PCR for other trypanosomes, but positive to T. vivax which yielded an expected amplicon of 175 bp. However, the Northern Guinea Savannah sample (Kaduna sample) was negative to T. vivax species-specific PCR. These two tests confirmed that the isolates used in the study were pure T. vivax and T. congolense.
Observation on the RAPD-PCR of T. vivax and T. congolense
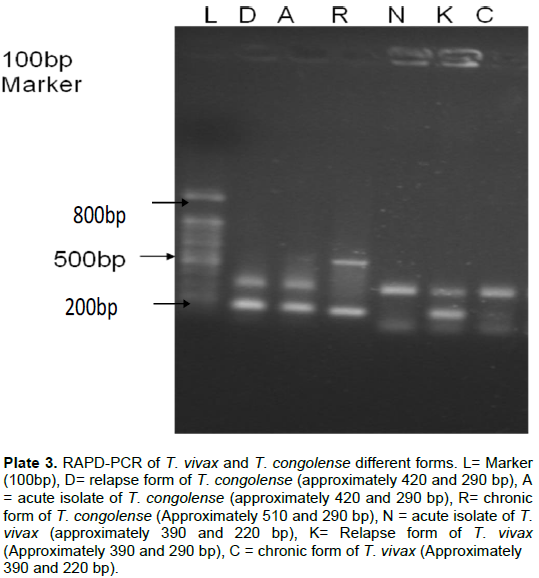
The experiment aimed at checking whether there was alteration of the DNA through change in bands of the T. vivax and the T. congolense at different stages (acute, relapse and chronic) of the disease or not. The results of the experiment revealed that the amplicons of the acute (N) and chronic forms (C) of the T. vivax were the same, whereas that of the relapse form (K) was different.
However, in T. congolense, the acute (A) and the relapse forms (D) yielded bands of the same molecular sizes but the chronic form (R) had a different pattern
from the other two forms (Plate 3).
The Trypanosoma Genus specific and T. vivax species-specific PCR that the samples were subjected to confirm the species of the isolates to be pure T. vivax and T. congolense by having the same band sizes with other T. vivax and T. congolense from different parts of the world. It is known that Genus-specific Trypanosoma PCR can simultaneously detect multiple infections with T. vivax, T. congolense and T. brucei brucei in ruminants. The method was reported to have almost zero percent error in detecting and differentiating the different species and even subspecies and strains of trypanosomes (Hutchinson et al., 2007; Garcia et al., 2014).
The isolates were confirmed through the detection of bands of 750 and 175 bp for the primers used in amplifying the fragments from T. congolense and T. vivax, respectively. The results obtained from the RAPD-PCR shows that there may be genetic alteration in the field isolate as seen in the acute, relapse and chronic forms of the experimental disease, since the fragments seen with the field isolate (acute form) and chronic form (about 350 and 175 bp) were the same but differed from that of the relapse form (about 350 and 195 bp) for T. vivax. In T. congolense, the bands observed with the acute and relapse forms were the same and differed from the bands of the chronic form. The different bands may be as a result of new mutant formation that resulted from the use of drugs in this study. The difference in band weights of the three forms of the two isolates may be responsible for the differences in the pathogenicity of the parasites. The genetic alterations (change in bands) may also contribute to the widely reported antigenic variation in trypanosomosis (Zambrano et al., 2002; Pays, 2005; Deitsch et al., 2009). It is reported that species differ in the organization of their silent VSG archive (Greif et al., 2013); genomic analysis of the different stages of these infections assists in knowing the genes responsible for the changes in these band weights observed. It would have been very helpful if the amplicons obtained from the RADP-PCR were also sequenced to know the nucleotides that were altered, as this may assist in the possible production of DNA vaccine and drugs development that will knock off that nucleotide region for the control or prevention of this disease. The findings of this work may serve as a stepping stone to the discovery of a DNA vaccine against the disease.