ABSTRACT
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common worldwide enzymopathy with approximately 400 million individuals affected. This inherited disease is sex-linked recessive inheritance. The high prevalence of certain variants of G6PD in different populations and ethnic groups increases the likelihood of finding associations with other pathologies. Sickle cell disease and thalassemia are the most common pathologies associated with G6PD deficiency. The aim of this study was firstly to study the prevalence of glucose-6-phosphate dehydrogenase deficiency (A-376/202) by molecular analysis in homozygous sickle cell patients, and secondly to study the influence of this association on the clinical severity of the disease. In a cross-sectional study, 100 patients aged 15 years with homozygous sickle cell disease in the stationary phase regularly monitored in a National Center for Blood Transfusion were included over a six-month period stretching from September 2015 to February 2016. An EDTA sampling tube was taken from each patient for the study of hematological parameters and a molecular study for the detection of mutations 376 and 202. Clinical, epidemiological and biological variables were collected using a questionnaire. Data was analyzed using Epi-info 7.2. The results of the study showed that the variant A- characterized by a double mutation (376/202) was found with a frequency of 13% (13/100) with a clear male predominance (p ˂ 0.006). Variant A- was statistically significantly associated with cholelithiasis (p˂0.031). This study is of therapeutic interest since the recognition of G6PD-deficient sickle cell disease would make it possible to take adequate preventive measures with respect to the taking of oxidizing drugs.
Key words: Glucose-6-phosphate dehydrogenase (G6PD) deficiency, sickle cell disease, stationary phase, Sénégal.
Glucose-6-phosphate dehydrogenase (G6PD) is a key enzyme in the pentose phosphate pathway, catalyzing the oxidation of glucose-6-phosphate to 6-phosphogluconate with concomitant reduction of nicotinamide adenine dinucleotide phosphate (NADP) to nicotinamide adenine dinucleotide reduced phosphate
(NADPH,H). The hereditary enzymopathy results from a mutation on the gene carried by the X chromosome at position q28. Examination of variants of G6PD shows that, in most cases, the deficiency is related to the instability of the enzyme, which implies that amino acid substitutions in different locations can destabilize the enzyme molecule (Cappellini and Fiorelli, 2008). Different types of deficits in G6PD have been characterized. Of these types of deficiency, some have normal activity and others have reduced activity. The most common of those who have normal activity is type A, which is found with great frequency in subjects from Africa. This variant is due to a substitution of guanine by adenine at position 376 causing a change in the amino acid asparalgine (Asn) by aspartic acid (Asp) in position 126 on the peptide chain. However, a second mutation found on this same allele due to the substitution of adenine by guanine at position 202 corresponds to the variant A- of G6PD. This represents 30% of the African black population (Cotton, 2003). The instability of the protein, which is created in the A- type deficiency, reduces the half-life of the protein from 62 to 13 days and its activity is only 0.8% of that of a young red cells.
The G6PD deficit affects more than 400 million people worldwide (Gentilini, 1993). It is the most common enzymopathy in the human species (Joseph et al., 1993). The WHO has classified the different variants of G6PD into three classes according to enzymatic activity and clinical manifestations: class 1 characterized by enzyme deficiency associated with non-spherocytic chronic hemolytic anemia, class 2 characterized by a severely deficient enzymatic activity with less than 10% of normal activity and class 3 characterized by a moderate enzymatic deficiency whose activity is between 20 and 60% of normal. The international classification classifies G6PD A- in type III (Jolly 2000). The analysis of the literature devoted to the study of the frequency of G6PD shows that it varies from one population to another, which can be related to the frequency of genetic factors or behaviors in each population. This great variability is related to G6PD deficiency detection techniques. The enzymatic techniques, although widely used, have limits especially during the post-hemolytic periods characterized by a large recruitment of reticulocytes which are rich in G6PD. This could lead to a misinterpretation of results obtained by biochemical techniques. With these findings, it is believed that molecular methods, although recent and few, seem to give results different from those obtained with biochemical techniques, especially during periods of heterozygosity. Thus, using the molecular technique, a study on homozygous sickle cell patients was conducted.
The main objective of this study is to determine the prevalence of G6PD type A- deficiency in homozygous sickle cell patients and to check the influence of this deficiency on the clinical severity of the disease.
The study took place in the Clinical Hematology Services of the National Blood Transfusion Center, the Fann Biochemistry Department and the Molecular Biology Unit of the Parasitology Department of the Cheikh Anta Diop University of Dakar. It is concerned with stationary phase homozygous sickle cell patients aged 15 years and over regularly monitored in this center. The stationary phase is defined by the absence of complication and acute manifestation of sickle cell disease within 15 days prior to recruitment. Each patient received an EDTA sampling tube for the study of haematological parameters and the molecular study for the detection of mutations 376 and 202 by PCR. This was a simple random sample on a population of 1400 sickle cell patients regularly monitored in this center.
Inclusion criteria
The inclusion criteria were as follows: 15 years old and over; agreement to participate in the study by the patient with written and signed informed consent; no manifestation or acute complication of sickle cell disease within 15 days of recruitment; electrophoretic profile confirmed by electrophoresis of haemoglobin.
Exclusion criteria
Patients with other chronic conditions including those with incomplete medical records were excluded from this study.
Ethical considerations
Free and informed consent written and signed by all participants was obtained beforehand.
Hematological parameters
A venous blood sample was taken on an empty stomach and collected in a tube containing EDTA as anticoagulant. The blood count was done with an automatic counter Coulter ABX pentra type. The electrophoretic profile of hemoglobin was determined by capillary technique using the SEBIA Minicap Flex Piercing on fresh blood collected on EDTA tube or stored at 2 to 7°C for less than seven days. The following decision criteria were used:
1. Anemia was defined as hemoglobin (Hb) Ë‚ 11 g/dl and categorized according to the mean corpuscular volume (MCV): in macrocytic anemia (MCV ˃ 95 fl), normocytic (80 ≤ MCV ≤ 95 fl) and microcytic (MCV Ë‚ 80fl). It is considered severe if Hb was Ë‚ 7 g/dl, moderate if 7 g/dl ≤ Hb ≤ 9 g/dl and light if 9 g/dl ≤ Hb ≤ 11 g/dl. According to the mean hemoglobin content (MCT), ranging from 27 to 31 pg, hypochromic anemia (MCT Ë‚27 pg) and normochromic anemia (27 Ë‚ MCT Ë‚ 31 pg) are distinguished.
2. The normal platelet count between 200000 and 400000/mm3
3. The normal rate of white blood cells (WBC) between 40000 and 10000/mm3
Molecular study
Extraction of DNA by Qiagen kit (Qiagen, http://www.qiagen.com)
The use of this kit allowed extraction of the DNA from a small volume of blood (200 μl). After lysis of the red blood cells, 2 ml of the "FG2" buffer and 20 μl of "Qiagen Protease" are added to the white blood cell pellet. The mixture is vortexed 3 to 4 times for 5 s and then incubated for 10 min at 65°C. Protein digestion gave the mixture a green color. 2 volumes of absolute ethanol are then added to the mixture, which is then centrifuged for 3 min at 2000 rpm. The precipitated DNA is recovered, washed with 5 ml of 70% ethanol, dried and finally dissolved in the "FG3" buffer for 1 h at 65°C.
Control of the purity of the DNA extracted for agarose gel electrophoresis and transfer after genotyping of G6PD polymerase chain reaction-restriction fragment length polymorphism (PCR-RLFP)
To evaluate the quality of the DNA, electrophoresis was carried out on a 3% agarose gel in 0.5x TBE buffer (4.45 mM Tris, 4.45 mM boric acid, 0.1 mM EDTA: pH = 8). 5 μl of DNA are added to 1 μl of the following blue mixture (0.25% of Xylenecyanol, 0.25% of bromophenol blue and 30% of glycerol). After migration and visualization under UV, a "smear" shows degraded DNA. On the other hand, a sharp band of high intensity attests to a non-degraded state of the nucleic acid. The principle was to diagnose point mutations in exons 4 and 5 by standard PCR with two primer pairs. The amplification of exon 4 was done using 1 μg of genomic DNA with the primers 5'-GTGGCTGTTCCGGGATGGCCTTCTG-3 '(forward) and 5'-CTTGAAGAAGGGCTCACTCTGTTTTG-3 '(reverse) and that of exon 5 with the primers
5'- CAGTACGATGATGCAGCC-3 '(forward) and 5'- CAGGTAGAAGAGGCGGT-3’ (reverse) (Inqaba, http://www.inqababiotec.com.za)
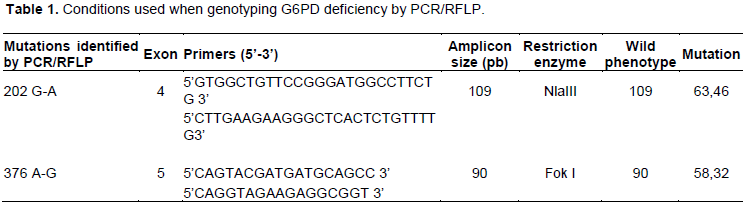
These primers are used under the following conditions: denaturation at 94°C for 5 min followed by 35 cycles of denaturation at 94°C for 1 min, elongation at 58°C for 1 min, extension at 72°C for 1 min and a final extension at 72°C for 10 min. By mixing the pair of primers with the human DNA, they were positioned at the level of their complementary sequences and by activating the Taq polymerase, each primer is extended in the direction 5 'to 3' of a sequence exactly complementary to the copied strand. The amplification product is then digested for 3 h at 37°C by restriction enzymes which recognize a specific restriction site. NlaIII (Inqaba, http://www.inqababiotec.com.za) for the 202G/A mutation and the 376A/G mutation by FokI (Inqaba, http://www.inqababiotec.com.za). This digestion was performed in a final product of 20 μL comprising: 7μl of amplification product; 2.5 μl of enzyme specific buffer; 0.5 μl of enzyme; 10 μl of water.
The digests were analyzed on a 3% agarose gel containing ethidium bromide. The conditions used for genotyping the G6PD deficiency were summarized in the following Table 1.
Figures 1 and 2, respectively illustrate the size of the fragments after amplification of exon 4 comprising the 202 G-A mutation and the exon 5 comprising the 376 A-G mutation as well as the digestion product in the presence or absence of mutations.
Data collection and statistical analysis
For each patient, the epidemiological, clinical, hematological and molecular data were collected on a survey card and then entered on a computer and analyzed using the CDC (Central of Control Disease) Epi-info 7.0 software, Atlanta, USA. The Student t-test was used to compare the means of the quantitative variables and the Chi2 test for the qualitative variables. The significance threshold for statistical tests was set at p Ë‚0.05.
Epidemiological characteristics of the study population at baseline
The average age of the patients was 28 ± 8.3 with extremes of 15 and 57 years. The age group most represented was between 15 and 25 years. There was a slight female predominance with a sex ratio of 0.75. the percentage decreased with age. Six patients were over the age of 46. Figure 3 illustrates the distribution of patients by sex.
Clinical characteristics
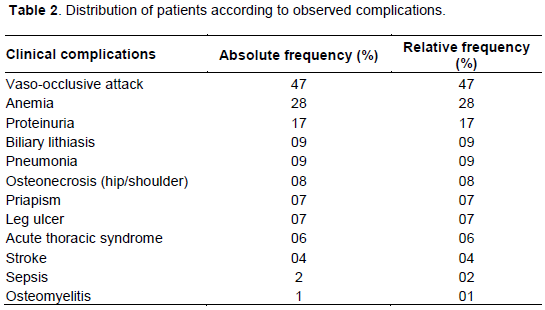
Complications were dominated by vaso-occlusive crisis 47 patients (47%) of the study population of which 38 had two seizures. Table 2 summarizes these complications.
Hematological characteristics
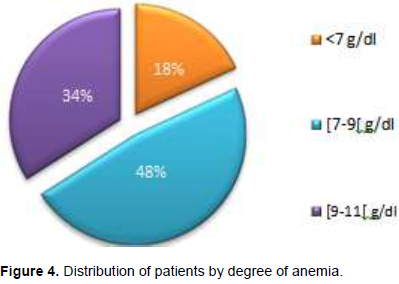
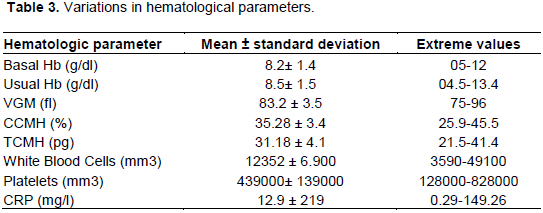
The baseline hemoglobin level was between 5 and 12 g/dl with a mean of 8 ± 1.4 g/dl. Mean corpuscular hemoglobin concentration (MCHC) and mean corpuscular hemoglobin MCH were 35.28 ± 3.46% and 31.18 ± 4.1 pg, respectively. Anemia was observed in 94% of patients. The degree of anemia was variable, 18% had severe anemia, 48% had moderate anemia and 34% had minor anemia. Figure 4 illustrates this distribution.
The mean state leukocytosis of 12345 ± 6900/mm3 with extremes ranging from 3500 to 49000/mm3. Hyperleukocytosis (WBC > 10000/mm3) was observed in 53% of patients. The mean platelet count was 439000 ± 139330/mm3 with extremes ranging from 180000 to 828 000/mm3. 47% had thrombocytosis. Mean hemoglobin fractions were 87.78% for hemoglobin S ± 8.19%, 2.72 ± 1.04% for hemoglobin A2 and 9.50 ± 8.34% for hemoglobin F, respectively. Table 3 reports the mean values of the hematological parameters of the patients.
Molecular characteristics
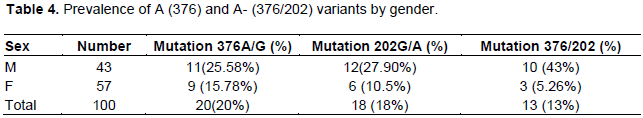
G6PD status was normal in 80% of patients with variant A. The frequency of heterozygosity was 15% and homozygosity 5%. For the variant A-, the G6PD status was normal in 87%. The frequency was 5.26% for women and 23% for men. The difference between men and women was statistically significant (p˂0.006). Table 4 summarizes the prevalence of A and A-mutations by sex.
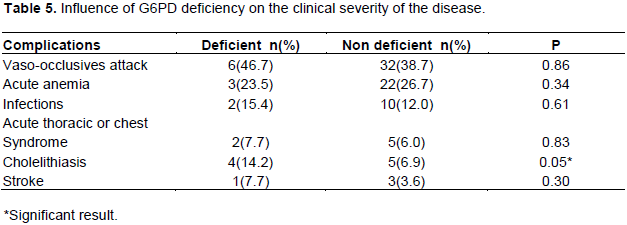
The relationship between clinical severity and G6PD deficiency varying A- shows that this deficit was statistically associated with cholelithiasis (p˂0.006). A relationship was also observed between the existence of this mutation and the baseline hemoglobin level, but the difference was not statistically significant (p = 0.08). No statistically significant difference was found with the number of vaso-occlusive attacks, anemia, transfusions, infectious complications and acute thoracic syndrome. Table 5 summarizes the influence of variant A- on the clinical severity of the disease.
Sickle cell anemia and G6PD deficiency are two abnormalities of the red blood cell responsible for
hemolytic anemia. Their association with the same patient deserves to be known in order to better adapt the care. The aim of this work was to study the prevalence of G6PD deficiency variant A- (3762/202) in homozygous sickle cell patients and the influence of this mutation on the clinical severity of patients. The enzymatic techniques, although widely used, have limits especially in homozygous sickle cell disease since the deficiency is often masked by permanent reticulocytosis. This could lead to a misinterpretation of results generated by biochemical techniques. It is for this reason that we have used the molecular method which makes it possible to identify the responsible defect and to predict the degree of clinical severity that the patient is experiencing.
The socio-demographic characteristics of this study population indicate that the mean age is 28 years with a sex ratio (M/F) of 0.75. These results are consistent with Shongo et al. (2015) study in the Democratic Republic of Congo, which showed a female predominance with a sex ratio of 0.8. Clinically, vaso-occlusive attacks dominated the symptomatology of sickle cell disease in 48% of cases. These findings correspond with the study conducted by Diagne et al. (2000), where vaso-occlusive attacks were the first clinical manifestation in terms of frequency (67%). Hematologically, the majority of patients were anemic (94%). This anemia is normochromic normocytic in 73% of cases. These data are in agreement with literature data which associates normochromic anemia to this condition (Nacoulma et al., 2014; Dahmani et al., 2016). Hyperleukocytosis was noted in 53% of patients with a mean of 12352 ± 6900/mm3. Similar results have been found by many authors: Nacoulma et al. (2014) in Mali and Dahmani et al. (2016) in Algeria. In this study, leukocytosis favours an inflammatory reaction that can increase the number of leucocytes because it correlates statistically significant with CRP (r = 0.72, p Ë‚ 0.01).
The results of molecular genotyping of G6PD revealed a prevalence of about 13% of the A-variant. This prevalence was similar to that described by Dolo et al. (2014) in Mali, which was 13.3% among the Dogon. However, it is relatively weaker than that described by Diop et al. (2000) in his study of 55 SS sickle cell patients with a prevalence of 32.7. This difference would be related to the criteria of inclusion of the patients but especially to the method of detection which was rather biochemical. The male prevalence found in this study has been described by many authors including Vizzi et al. (2016) in Venezuela, which found a prevalence of 5.5% in men as compared to 4.1% in women (p Ë‚ 0.007). Similar results were also obtained by Benkerrou et al. (2013) with respectively 16% of men versus 10% with women.
The comparison of the evolutionary profile between sickle cell deficient and non-deficient patients showed an absence of influence of the enzymopathy of the severity of the disease in these patients. Only Cholelithiasis was significantly associated with G6PD deficiency. Several authors have been interested in the aspect of the problem and the conclusions are divergent. For some, the G6PD deficiency would have a protective effect since the absence of enzymatic activity could contribute to the premature disappearance of red blood cells, which would lead to the circulation of a new generation of circulating young cells. In a study on homozygous sickle cell patients, Ahmed and Ibrahim (2002) suggested that those with G6PD deficiency have significantly less vaso-occlusive attacks than non-deficient patients. This suggests that co-inheritance of G6PD deficiency in sickle cell patients reduces vaso-occlusive attacks and promotes the prognosis of disease. In addition to compensatory medullary regeneration, the hypothesis put forward to explain this absence of influence is the less severe type A- which is predominant in black. Most studies confirm our results and do not find any influence of this variant on the severity of the disease (Bouanga et al., 1998; Nouraie et al., 2010). However, the results should not obscure the fact that the combination of sickle cell disease and G6PD deficiency presents obvious potential risks. Indeed, most of the drugs used against sickle cell disease are oxidizing and therefore likely to cause haemolysis in the deficient. Several authors have demonstrated that oxidative stress during infection and the use of analgesics during vaso-occlusive attacks contribute to increased severity of hemolysis in patients with G6PD deficiency (Cappellini and Fiorelli, 2008; Nouraie et al 2010). This phenomenon seems to be confirmed with the association G6PD deficiency and Cholelithiasis in this study. Longitudinal study with a larger sampling would be needed to confirm this association. This study was limited in our inability to screen the type of haplotype present in our patients as these are known to impact clinical severity of the disease.
Finally, it can be said that the frequency of the erythrocyte G6PD deficiency type A-376/202 is comparable to that found in some African regions, particularly in Mali. This study also revealed that the deficit does not influence the clinical severity of the disease. These results, however, should not obscure the fact that the association of G6PD-deficiency sickle cell disease presents potential risks because of the oxidative drugs used. It is therefore useful to detect G6PD deficiency in sickle cell patients in order to deduce preventive and therapeutic measures.
The authors have not declared any conflict of interests.
The authors are thankful to all the patients at the National Blood Transfusion Center for their commitment in this study. Thanks also go to the various services that helped in the study of the patients.
REFERENCES
|
Ahmed SG, Ibrahim VA (2002). Clinical significance of glucose-6-phosphate dehydrogenase deficiency in Nigerian patients with sickle cell disease. Nigerian Postgraduate Medical Journal 9(4):181-185.
|
|
|
|
Benkerrou M, Alberti C, Couque N, Haouari Z (2013). Impact of glucoseâ€6â€phosphate dehydrogenase deficiency on sickle cell anaemia expression in infancy and early childhood: A prospective study. British Journal of Haematology 163(5):646-654.
Crossref
|
|
|
|
|
Bouanga JC, Mouélé R, Préhu C, Wajcman H, Feingold J (1998). Glucose-6-phosphate dehydrogenase deficiency and homozygous sickle cell disease in Congo. Human Heredity 48(4):192-197.
Crossref
|
|
|
|
|
Cappellini MD, Fiorelli GE (2008). Glucose-6-phosphate dehydrogenase deficiency. The lancet 371(9606):64-74.
Crossref
|
|
|
|
|
Cotton F (2003). Contribution au diagnostic biochimique des maladies héréditaires de l'hémoglobine. Thèse Doctorat Université Libre de Bruxelles. Available at :
View
|
|
|
|
|
Dahmani F, Saoud B, Jafaar K, Woumki A (2016). Etude de l'hémogramme dans la drépanocytose homozygote: à propos de 87 patients. The Pan African Medical Journal 25:240.
Crossref
|
|
|
|
|
Diagne I, Ndiaye O, Moreira C, Signate-Sy H, Camara B, Diouf S, Diack-Mbaye A, Ba M, Sarr M, Sow D, Fall M (2000). Les syndromes drépanocytaires majeurs en pédiatrie à Dakar (Sénégal). Archives de Pédiatrie 7(1):16-24.
Crossref
|
|
|
|
|
Diop S, Thiam D, Sene A, Cisse M, Fall K, Toure Fall AO, Sow O, Diakhate L (2000). Association drépanocytose-déficit en G-6-PD:-prévalence et influence sur le profil évolutif. Médecine d'Afrique Noire 47(7):321-326.
|
|
|
|
|
Dolo A, Maiga B, Guindo A, Diakité SA, Diakite M, Tapily A, Traoré M, Sangaré B, Arama C, Daou M, Doumbo O (2014). Fréquence du déficit en glucose-6-phosphate déshydrogénase (A-376/202) dans trois groupes ethniques vivant en zone d'endémie palustre au Mali Frequency of Glucose-6-phosphate dehydrogenase deficiency (A-376/202) in three Malian ethnic groups. Bulletin de la Société de Pathologie Exotique 107(3):165-170.
Crossref
|
|
|
|
|
Gentilini M (1993). Enzymopathie érythrocytaire. Médecine Tropicale, Flammarion Eds, Paris, France. pp. 532-537.
|
|
|
|
|
Jolly D (2000). Le déficit en G6PD: une affection génétique fréquente et mal connue: un cas d'école en santé publique. Flammarion médecine-sciences. Available at:
View
|
|
|
|
|
Joseph R, Ho LY, Gomez JM, Rajdurai VS, Sivasankaran S, Yip YY (1999). Mass newborn screening for glucose-6-phosphate dehydrogenase deficiency in Singapore. The Southeast Asian Journal of Tropical Medicine and Public Health 30(2):70-71.
|
|
|
|
|
Nacoulma E, Sakande J, Kafando E, Kpowbié ED, Guissou IP (2006). Profil hématologique et biochimique des drépanocytaires SS et SC en phase stationnaire au Centre Hospitalier National Yalgado Ouedraogo de Ouagadougou. Mali Medical 21(1):8-11.
|
|
|
|
|
Nouraie M, Reading NS, Campbell A, Minniti CP, Rana SR, Luchtmanâ€Jones L, Kato GJ, Gladwin MT, Castro OL, Prchal JT, Gordeuk VR (2010). Association of G6PD202A, 376G with lower haemoglobin concentration but not increased haemolysis in patients with sickle cell anaemia. British Journal of Haematology 150(2):218-225.
Crossref
|
|
|
|
|
Shongo M, Mukuku O, Mutombo A, Loubala T (2015). Hematological and nutritional profile of SS homozygous sickle cell aged 6-59 months in Lubumbashi, Democratic Republic of Congo. Pan African Medical Journal 21(1):276.
|
|
|
|
|
Vizzi A, Bastisdas G, Hidalgo M, Colman L, Perez HA (2016). Prevalence and molecular characterization of G6PD deficiency in two Plasmodium vivax endemic areas in Venezuela: predominance of the African A-202A/376G variant. Malaria Journal 15(1):19.
Crossref
|
|