Full Length Research Paper

ABSTRACT
Trichloroethylene (TCE), a common water pollutant linked to Parkinson’s Disease (PD), induces dopaminergic neurodegeneration. L-Theanine (L-Th) was explored as a potential treatment for TCE-induced PD due to its previously elucidated neuroprotective properties. Cell viability, cytotoxicity, and cell density were evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay (n=8), lactate dehydrogenase (LDH) assay (n=4), and cell image analysis (n=6), respectively. GT1-7 and SK-N-SH cells served as dopaminergic and neuronal cell models, respectively. In GT1-7 cells, L-Th 600 μM diminished TCE 1000 μM-induced cell death and TCE 1000 μM-induced LDH release by 81% (p<0.001) and 38% (p<0.001), respectively, after 48 h. L-Th also did not significantly impact LDH leakage in healthy GT1-7 cells. In SK-N-SH cells, L-Th 600 μM attenuated TCE 100 μM’s neurodegenerative effects by increasing cellular density and cellular area by 118% (p<0.01) and 170% (p<0.001), respectively, after 24 h. L-Th’s mitigation of TCE’s neurotoxic and neurodegenerative effects in dopaminergic neurons can prevent dopaminergic neurodegeneration: linked to PD onset. L-Th’s ability to preserve healthy GT1-7 cells indicates that L-Th not neurotoxic in vitro. This research marks the identification of the first potential treatment for TCE-induced PD. Future investigations should explore the mechanism of L-Th and TCE’s interactions.
Keywords: Trichloroethylene, environmental toxin, L-Theanine, Parkinson’s disease, neurodegeneration.
INTRODUCTION
Parkinson's Disease (PD), a late-onset neurodegenerative disorder, currently affects 10 million people worldwide; more specifically in the US alone, PD affects 1 million people with an additional 60,000 people diagnosed annually (Ball et al., 2019; DeMaagd and Phillip, 2015). PD pathogenesis is attributed to two main biological processes: the degradation of dopaminergic neurons and accumulation of alpha-synuclein (a-syn) in the brain (DeMaagd and Phillip, 2015; Emamzadeh and Surguchov, 2018). These processes work synergistically to induce the characteristic lack of dopamine in PD patients; a shortage that is responsible for PD-induced motor degeneration including tremors, bradykinesia, and impaired coordination (DeMaagd and Phillip, 2015; Emamzadeh and Surguchov, 2018).
Scientists are currently aware of two classifications of PD: familial and sporadic (Inamdar et al., 2007; Muñoz et al., 2001). Familial PD, PD initiated by genetic factors, accounts for a mere 15% of all PD cases while Sporadic PD, PD initiated by environmental factors, accounts for a substantial 85% of all PD cases (Inamdar et al., 2007; Muñoz et al., 2001). This has led to numerous research publications detailing the alternative pathways in which sporadic PD progresses (Braak et al., 2003; Braak et al., 2003; Rietdijk et al., 2017; Yulug et al., 2019). Braak et al. (2001) theorized one of the most widely accepted hypotheses in PD research: Braak’s hypothesis (Braak et al., 2003). Braak’s hypothesis states that an ingested environmental toxin could initiate PD onset through alpha-synuclein aggregation in gastrointestinal cells which would allow alpha-synuclein to travel to the brain via the gut-brain axis: the vagus nerve (Braak et al., 2003). Similarly, inhaled environmental toxins can initiate PD onset through alpha-synuclein aggregation in the olfactory tract which can travel to the brain via the olfactory bulb (Rietdijk et al., 2017). These monumental studies set forth a cascade of research investigating the interactions between environmental toxins in living organisms.
Trichloroethylene and Parkinson’s disease
One such environmental toxin is trichloroethylene (TCE), a commonly used industrial solvent (Doherty, 2000). TCE’s widespread use in factories has led to its improper disposal and infiltration into over 30% of USA ground and surface water drinking supplies (Doherty, 2000; Zaheer and Slevin, 2011). TCE has also become a prevalent air pollutant (Doherty, 2000). In accordance with Braak’s hypothesis, TCE-induced PD is hypothesized to progress in living organisms through the ingestion of TCE-contaminated drinking water and inhalation of TCE-contaminated air through the gastrointestinal and olfactory tract, respectively (Braak et al., 2003; Khare et al., 2018; Rietdijk et al., 2017; Yulug et al., 2019). More importantly, TCE is also able to infiltrate the blood-brain barrier (BBB) allowing for accelerated neurodegeneration and PD onset (Bonvallot et al., 2010).
TCE acts as a complex I mitochondrial inhibitor leading to mitochondrial dysfunction, reduced ATP levels, disrupted Ca+2 homeostasis, and ultimately cell death (Liu et al., 2010; Zaheer and Slevin, 2011). Previous in vivo studies have demonstrated TCE as a neurotoxic compound linked with the onset of PD (Liu et al., 2010; Zaheer and Slevin, 2011). Fisher 344 rats exposed to TCE exhibited dopaminergic neurodegeneration in the substantia nigra pars compacta and a-syn expression in the dorsal motor nucleus of the vagus nerve (Liu et al., 2010; Zaheer and Slevin, 2011). TCE also upregulated the expression of reactive oxygen species (ROS) leading to elevated oxidative stress in the brain: linked to neuroinflammation and a-syn aggregation (Liu et al., 2010; Sharma and Nehru, 2017).
TCE’s linkage with PD has led to the slight reduction in its usage; however, unlike other environmental toxins implicated in PD pathogenesis, TCE is still legally permitted for use and still poses a threat to human health (Krasnic, 2017). According to a 2016 study, over 171,000,000 lbs of TCE were still manufactured in the USA in 2015 despite TCE’s notorious neurotoxic properties (Krasnic, 2017). Minimal TCE bioremediation efforts coupled with TCE’s inability to vaporize in groundwater has led to its increased environmental presence (Russell et al., 1992). Despite TCE’s potent neurotoxicity and linkage with PD, scientific literature has not identified any explicit treatment for TCE-induced PD (Figure 1).
Dopamine and Parkinson’s disease
One of the primary biomarkers observed in PD patients is the degeneration of dopaminergic neurons (DeMaagd and Phillip, 2015; Emamzadeh and Surguchov, 2018). Dopaminergic neurodegeneration is singlehandedly responsible for a majority of PD-related motor degeneration (Triarhou, 2013). PD patients can expect to lose up 60-80% of all dopaminergic neurons throughout PD progression leading to the depletion of dopamine within the brain (Cheng et al., 2010). Due to dopamine’s crucial role in facilitating bodily movements, decreased dopamine levels lead to the progression of PD’s most common symptoms: motor dysfunction in the hands and muscles (Triarhou, 2013).
L-Theanine as a treatment for trichloroethylene-induced Parkinsonism
Recent literature has implicated green tea compounds in the treatment for PD (Deb et al., 2019; Chen et al., 2018). A large amount of green tea’s neuroprotective attributes can be traced back to L-Theanine (L-Th), a neuroprotective amino acid analog that occurs naturally in green tea leaves (Camellia sinensis) (Deb et al., 2019; Chen et al., 2018).
L-Th’s unique ability to lessen the effects of neurotoxic compounds through downregulating heme oxygenase allows it to significantly reduce SH-SY5Y cell death caused by other environmental toxins such as rotenone and dieldrin (Cho et al., 2008). L-Th can also upregulate dopamine levels in the striatum, hippocampus, and hypothalamus which may allow for the reversal of PD-induced dopamine deficiency and motor impairment (Yokogoshi and Terashima, 2000). Although L-Th is not a known antioxidant, it reduces intracellular reactive oxygen species (ROS) through enhancing cellular antioxidant defense mechanisms (Ben et al., 2015; Deb et al., 2019). L-Th has also shown the potential to reduce the expression of inflammatory cytokines and increase the motor functionality of motor-impaired organisms (Deb et al., 2019; Li et al., 2016; Thangarajan et al., 2014).
In accordance with L-Th’s previously stated neuroprotective attributes, L-Th is an excellent candidate for treating TCE-induced PD. L-Th’s ability to reduce neuronal apoptosis along with the upregulation of free dopamine in the brain can mitigate TCE’s neurotoxic effects on dopaminergic neurons and dopamine levels (Cho et al., 2008; Yokogoshi and Terashima, 2000). L-Th’s upregulation of cellular antioxidant molecules may also attenuate TCE-induced oxidative stress (Ben et al., 2015; Deb et al., 2019). Additionally, L-Th’s capability of passing the blood-brain barrier via the leucine-preferring transport system further optimizes L-Th as a potential treatment for TCE-induced PD by minimizing the invasiveness of its potential application in vivo (Chen et al., 2018) (Figure 2).
Purpose
Although TCE-induced PD clearly poses an imminent threat on communities with contaminated drinking water, scientific literature has failed to identify any specific treatment for TCE-induced PD. Therefore, the purpose of this research was to identify L-Th as an effective treatment for TCE-induced PD through the visualization of cell viability, cytotoxicity, cell density and cell morphology in a Parkinson’s disease cell model.
MATERIALS AND METHODS
Cell lines
In this study, dopaminergic neuron models and neuronal cell models were used due to their relevance in PD studies. GT1-7 mouse hypothalamic cells (ATCC; USA) were grown in Modified Eagle's Medium (ATCC; USA) with 10% fetal bovine serum (Invitrogen, USA) at 37°C and 5% CO2. GT1-7 mouse hypothalamic cells served as a dopaminergic neuron model to assess L-Th and TCE’s impacts on cell viability and cytotoxicity. SK-N-SH human neuroblastoma cells (ATCC; USA) were grown in Modified Eagle’s Medium (ATCC; USA) with 10% fetal bovine serum (Invitrogen, USA) at 37°C and 5% CO2. SK-N-SH human neuroblastoma cells served as a neuronal cell model to assess L-Th and TCE’s impacts on cell density.
MTT cell viability assay
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay was conducted to observe TCE and L-Th’s impact on cell viability. MTT cell viability assay was used to quantify the number of metabolically viable cells by observing the amount of purple formazan formed after the addition of the metabolizable MTT reagent. TCE and L-Th were initially added to cells in a 96-well plate and incubated at 37°C and 5% CO2 for 24 h or 48 h. After the initial incubation, 10 μl of MTT reagent was added to each well and incubated again at 37°C and 5% CO2 for 3 h allowing viable cells to convert the MTT reagent into a purple formazan. Next, 70 μl of Dimethyl sulfoxide was added to each well to solubilize the plasma membrane of the cells allowing for the release of the purple formazan into the extracellular media. This 96 well plate was then placed into a microplate reader (Bio-Rad, USA) and read at 595 nm. This data was then loaded into the Microplate Manager 4.0 software (Bio-Rad, USA). MTT cell viability assay (n=8) was conducted at 24 and 48 h intervals to assess the impacts of TCE and L-Th on cell viability.
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) assay was conducted to observe TCE and L-Th’s impact on cytotoxicity via plasma membrane damage. LDH cytotoxicity assay was used to quantify the cytotoxicity of chemicals by observing LDH release into the extracellular region. This LDH release could be measured through the addition of the LDH substrate that catalyzes the conversion of LDH into red formazan. TCE and L-Th were initially added to cells in a 96-well plate and incubated at 37°C and 5% CO2 for 24 h or 48 h. After the initial incubation, 50 μl of cell media was transferred into a separate 96-well plate. Next, 50 μl of LDH substrate was added to each well and placed away from light for 30 min allowing for the LDH to escape into the extracellular region in damaged cells where it is converted into a red formazan. Next, 50 μl of LDH assay stop solution was added to each well to stop the chemical reaction occurring. This 96 well plate was then placed into a microplate reader (Bio-Rad, USA) and read at 490 and 655 nm. This data was then loaded into a Microplate Manager 4.0 software (Bio-Rad, USA). LDH cytotoxicity assay (n=4) was conducted at 24 and 48 h intervals to assess the impacts of TCE and L-Th on cytotoxicity.
Cell image analysis
Cell image analysis was conducted to observe the effects of TCE and L-Th on cellular density. After TCE and L-Th were added to cells, the 6-well plate was incubated at 37°C with 5% CO2 for 24 h Next, all cell media was removed and 1000 μl of Hema-3 fixative was added into each well fixing the cells. The Hema-3 fixative was then taken out of each well and replaced with 1000 μl of hematoxylin for 2 min to stain the cells. After removing the hematoxylin, each well was rinsed twice with 1000 μl of distilled water. The cells were then observed under x4 and x40 magnifications through an imaging microscope using the AmScope software. All images were quantified through the use of the ImageJ software for cellular density and cellular area. Cell image analysis (n=6) was conducted on a 24 h interval to assess the impacts of TCE and L-Th on cell morphology.
Statistical analysis
All experiments were analyzed for significance using the GraphPad Prism 8.0 software (GraphPad Software; San Diego, CA) through a one-way analysis of variance (ANOVA) test to determine for significance among the results. A post-hoc Tukey test was then used to determine significance between specific treatment groups. P-values of less than or equal to 0.05 were considered significant. All error bars represent a 5% confidence interval.
RESULTS
Effects of TCE and L-Th on GT1-7 cell viability after 24 h and 48 h (Dopaminergic Neuron Model)
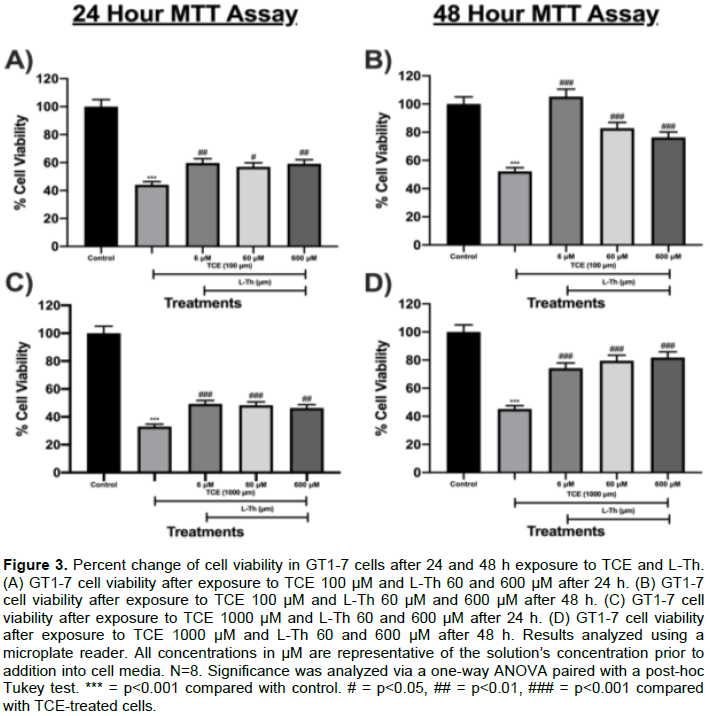
In this experiment, the effects of L-Th and TCE on GT1-7 cell viability were explored using MTT cell viability assay. L-Th was successful in increasing GT1-7 cell viability in TCE-treated cells. TCE 100 μM decreased cell viability by 56% after 24 h (p<0.001; Figure 3A) and 48% after 48 h (p<0.001; Figure 3B) compared to control. However, L-Th6, 60 and 600 μM increased cell viability in TCE 100 μM-treated cells by 35% (p<0.01; Figure 3A), 29% (p<0.05; Figure 3A), and 34% (p<0.01; Figure 3A), respectively, after 24 h. L-Th 6, 60, and 600 μM also increased cell viability in TCE 100 μM-treated cells by 101% (p<0.001; Figure 3B), 58% (p<0.001; Figure 3B), and 46% (p<0.001; Figure 3B), respectively, after 48 h. TCE 1000 μM decreased cell viability by 67% after 24 h (p<0.001; Figure 3C) and 55% after 48 h (p<0.001; Figure 3D) compared to control. However, L-Th 6, 60 and 600 μM increased cell viability in TCE 1000 μM-treated cells by 49% (p<0.001; Figure 3C), 46% (p<0.001; Figure 3C), and 40% (p<0.01; Figure 3C), respectively, after 24 h. Similarly, L-Th 6, 60 and 600 μM increased cell viability in TCE 1000 μM-treated cells by 64% (p<0.001; Figure 3D), 76% (p<0.001; Figure 3D), and 81% (p<0.001; Figure 3D), respectively, after 48 h.
The effects of TCE and L-Th on GT1-7 Cell LDH release after 24 h and 48 h (Dopaminergic Neuron Model)
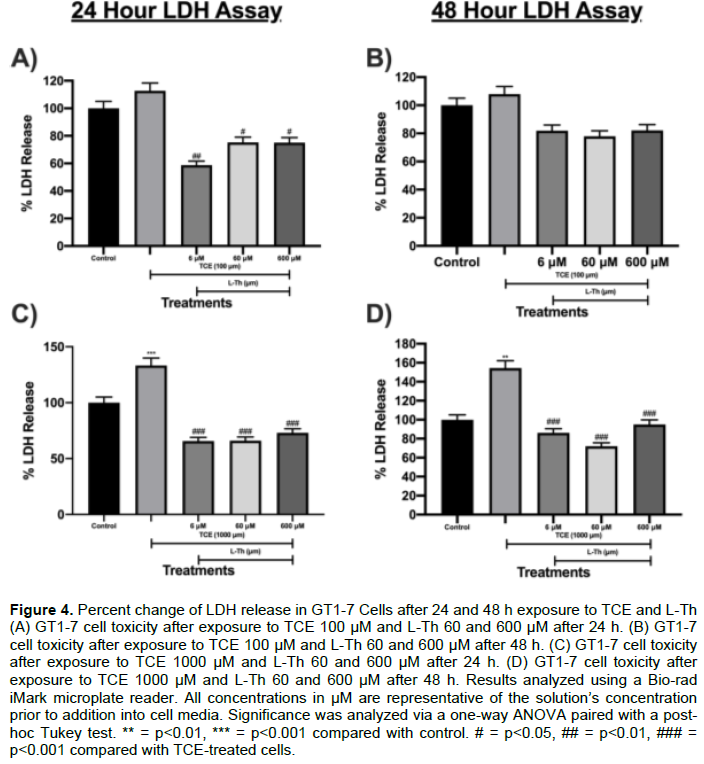
In this experiment, the effects of TCE and L-Th on GT1-7 cytotoxicity via plasma membrane degradation were analyzed using LDH assay. L-Th was successful at impeding TCE-induced LDH release from cells. TCE 100 μM did not significantly impact LDH release after 24 h or 48 h compared to control. Nonetheless, upon L-Th addition, L-Th 6, 60 and 600 μM decreased TCE 100 μM-induced LDH release by 48% (p<0.01; Figure 4A), 33% (p<0.05; Figure 4A), and 33% (p<0.05; Figure 4A), respectively, after 24 h. Despite this, L-Th 6, 60 and 600 μM didn’t significantly impact LDH release after 48 h (Figure 4B). TCE 1000 μM increased LDH release by 33% after 24 h (p<0.001; Figure 4C) and 53% after 48 h (p<0.01; Figure 4D) compared to control. However, L-Th 6, 60 and 600 μM attenuated TCE-induced LDH release by 51% (p<0.001; Figure 4C), 50% (p<0.001; Figure 4C), and 45% (p<0.001; Figure 4C), respectively, after 24 h. Likewise, L-Th 6, 60 and 600 μM mitigated TCE-induced LDH release by 44% (p<0.001; Figure 4D), 53% (p<0.001; Figure 4D), and 38% (p<0.001; Figure 4D), respectively, after 48 h.
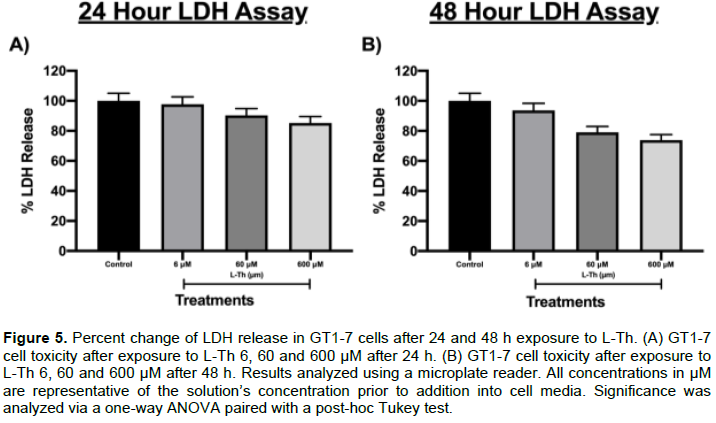
The effects of L-Th on untreated GT1-7 cell LDH release after 24 and 48 h (Dopaminergic Neuron Model)
L-Th’s cytotoxic effects on untreated GT1-7 cells were also assessed using the LDH assay to determine possible adverse reactions from L-Th administration on healthy cells. L-Th 6, 60 and 600 μM did not significantly impact LDH release from GT1-7 after 24 h (Figure 5A). Similarly, L-Th 6, 60 and 600 μM did not significantly impact LDH release from GT1-7 after 48 h (Figure 5B).
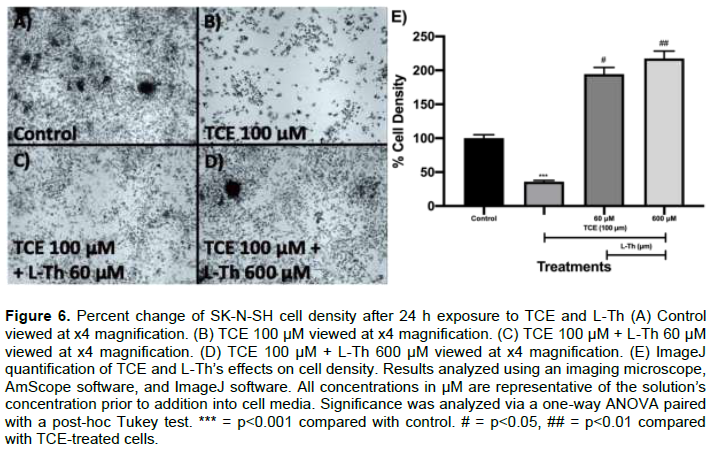
The effects of TCE and L-Th on SK-N-SH cell number and density after 24 h (Neuronal Cell Model)
Cell image analysis viewed at a x4 magnification was conducted to analyze the effects of TCE and L-Th on SK-N-SH cell density and neurodegeneration. Untreated SK-N-SH cells (control) exhibited a high cell density when examined visually (Figure 6A). Upon TCE 100 μM addition, cell density decreased dramatically when visually examined (Figure 6B). Upon ImageJ quantification, TCE 100 μM decreased cell density by 64% (p<0.001; Figure 6E) after 24 h. However, both L-Th 60 μM and 600 μM increased visual cell density in a dose-dependent manner after 24 h (Figure 6C and D). After ImageJ quantification, L-Th 60 and 600 μM increased cell density by 95% (p<0.05; Figure 6E) and 119% (p<0.01; Figure 6E), respectively, after 24 h.
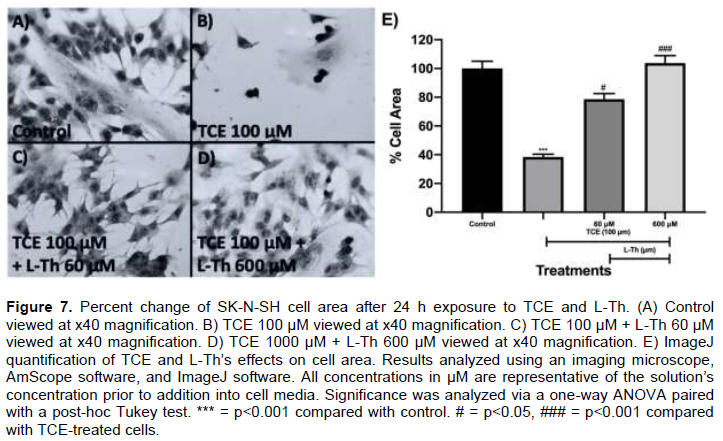
TCE and L-Th’s impact on SK-N-SH cell morphology after 24 h (Neuronal Cell Model)
Cell image analysis viewed at x40 magnification was conducted to assess TCE and L-Th’s impacts on SK-N-SH cell morphology. In the control, SK-N-SH cells demonstrated high cellular area (Figure 7A). However, TCE 100 μM-treated cells demonstrated diminished cell area after 24 h (Figure 7B). Upon ImageJ quantification, TCE 100 μM-treated cells exhibited a 62% reduction in cellular area (p<0.001; Figure 7E). Upon L-Th addition, both L-Th 60 μM and 600 μM drastically increased cellular area compared to TCE 100 μM-treated cells after 24 h (Figure 7B, C, and D). After ImageJ qunatification, L-Th 60 and 600 μM increased cellular area by 104% (p<0.05; Figure 7E) and 170% (p<0.001; Figure 7E), respectively, after 24 h.
DISCUSSION
Deb et al. (2019), hypothesized the utilization of L-Th as a potential treatment for PD as a result of L-Th’s previously elucidated neuroprotective effects (Deb et al., 2019). MTT assay allows for the observance of TCE and L-Th’s effects on cell viability in terms of mitochondrial function; therefore, L-Th’s mitigation of TCE-induced cell death suggests that L-Th may prevent TCE-induced cell death through the preservation of mitochondrial function (Mahajan et al., 2012). TCE’s neurotoxic effects on GT1-7 cells is supported by previous literature (Gash et al., 2008; Goldman, 2010; Guehl et al., 1999; Zaheer and Slevin, 2011). Previous literature indicates that TCE induces cellular death through the inhibition of complex I mitochondrial structure, likely through its selective binding of the ubiquinone site within the complex I structure (Gash et al., 2008). This can lead to the PD hallmarks: mitochondrial dysfunction, upregulated Ca+2, mitochondrial lysis, and ultimately cell death (Brookes et al., 2004; Gash et al., 2008; Sharma et al., 2017). Increased intracellular Ca+2 levels can lead to the mitochondrial release of pro-apoptotic factors such as cytochrome c (Brookes et al., 2004). L-Th was able to mitigate TCE’s toxic effects at 6, 60 and 600 μM. Previous literature indicates that L-Th upregulates mitochondrial efficiency by increasing mitochondrial transmembrane potential and complex II mitochondrial activity which can attenuate TCE’s suppression of mitochondrial function and efficiency (Ben et al., 2015; Chen et al., 2018; Deb et al., 2019). L-Th may also reduce TCE-induced Ca+2 expression by suppressing increased Ca+2 entry into neurons through its selective inhibition of ionotropic receptors and metabotropic receptor type I (Deb et al., 2019). Furthermore, TCE-induced mitochondrial lysis is also responsible for the release of ROS into the cytoplasm leading to oxidative stress (Brookes et al., 2004). Although L-Th is not an antioxidant, it has been well documented that L-Th upregulates cellular antioxidant molecules such as glutathione (Deb et al., 2019). In one study, L-Th also upregulated the formation of anti-stress molecules, PLC-β1 and γ1, in rat cerebral cortical neurons (Deb et al., 2019). This may lead to the reduction of TCE-induced oxidative stress and cytotoxicity.
LDH assay provides insight on TCE-induced cytotoxicity in terms of plasma membrane integrity; because of this, L-Th’s attenuation of TCE-induced LDH release suggests that L-Th may prevent TCE-induced cytotoxicity through the protection of cell membrane integrity (Chan et al., 2013). TCE’s destruction of cellular membranes likely results from TCE’s ability to induce high levels of lipid peroxidation across various cell lines (Channel et al., 1998; Zhu et al., 2005). In turn, lipid peroxidation affects phospholipid interactions and cellular membrane integrity (Catalá and Díaz, 2016). However, L-Th’s ability to upregulate glutathione leading to decreased lipid peroxidation may be the cause of L-Th-induced membrane preservation in TCE-treated cells. L-Th also did not exhibit any cytotoxic effect towards healthy GT1-7 cells suggesting that L-Th would ameliorate TCE-induced toxicity in a dopaminergic neuron model without harming healthy neurons; a characteristic important in PD treatments as current treatments (levodopa), induce neurotoxicity to untreated neurons in vitro (Ziv et al., 1997).
L-Th’s mitigation of TCE-induced cell death and cytotoxicity demonstrated in the MTT assay and LDH assay, respectively, indicate that L-Th preserves both mitochondrial function and plasma membrane integrity in the presence of TCE. TCE’s reduction of cell density and neuronal cell agglomeration is supported by previous studies as previous literature has indicated that TCE metabolite, 1-Trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo), rushes neuroblastoma cells through the cell cycle leading to incomplete chromosome formation, improper chromosome alignment, and improper pre-mitotic preparations (Sharma et al., 2017). This rapid cell division subsequently leads to small and underdeveloped post-mitotic daughter cells (Sharma et al., 2017). This is explanatory for the diminished cellular area found in TCE-treated cells. Furthermore, TaClo also inhibits the synthesis of prostaglandin E2 (PGE2) through the inhibition of cyclooxygenase-2 (Sharma et al., 2017). A previous study elucidated PGE2’s vital role in neuron density and neurite growth in the NSC-34 neuronal cell line (Nango et al., 2017). However, L-Th in 6, 60, and 600 μM significantly increased neuronal cell density. Previous research indicates that L-Th prevents cancer cell lines from entering the later stages of the cell cycle through its selective inhibition of GLAST and GLT-1 receptors that facilitate the uptake of cellular nutrients which may mitigate TaClo’s detrimental effect of rushed cell division (Saqcena et al., 2013; Sugiyama et al., 2001). Previous literature also suggests that L-Th upregulates the expression of PGE2 in gastrointestinal cells through enhanced cyclooxygenase-2 expression which may counter TaClo’s downregulation of PGE2 and promote neurogenesis (Chatterjee et al., 2014).
CONCLUSION
In this study, L-Th impeded the progression of PD in vitro through its hindrance of the most prominent PD biomarkers such as dopaminergic neurodegeneration, dopaminergic cytotoxicity, reduced cell density, and reduced cell area. Thus, this study presents the first specific treatment candidate for TCE-induced PD in scientific literature.
L-Th’s novel inhibition and prevention of PD pathogenesis targets the underlying conditions inducing PD onset instead of temporarily alleviating PD symptoms. Although L-Th was identified as a possible treatment for TCE-induced PD, there are several limitations to this research. Although inferences can be made according to previous scientific literature, L-Th’s specific mechanism of protection in TCE-treated cells was unconfirmed. Additionally, L-Th’s promise as a treatment for TCE-induced PD has yet to be confirmed in vivo.
In this study, L-Th was successfully identified as a possible treatment for TCE-induced PD. The limitations in this research should be investigated in future studies to further confirm L-Th’s ability to inhibit PD pathogenesis. Specifically, the mechanism in which L-Th acts on TCE-treated cells should be investigated using a multi-tiered approach utilizing molecular docking, enzyme-linked immunosorbent assay (ELISA) assay, and a western blot analysis to identify specific cellular pathways in which L-Th operates and assess cellular protein expression in TCE-treated cells.
CONFLICT OF INTERESTS
The author has not declared any conflict of interests.
ACKNOWLEDGEMENT
The author would like to express his gratitude for the mentorship received from Dr. Wei Zhu during this research.
REFERENCES
|
Ball N, Teo W, Chandra S, Chapman J (2019). Parkinson's Disease and the Environment. Frontiers in Neurology, Volume 10. |
|
|
Ben P, Zhang Z, Xuan C, Sun S, Shen L, Gao Y, Cao X, Zhou Y, Lan L, Yin Z, Luo L (2015). Protective Effect of l-Theanine on Cadmium-Induced Apoptosis in PC12 Cells by Inhibiting the Mitochondria-Mediated Pathway. Neurochemical Research 40(8): 1661-1670. |
|
|
Bonvallot N, Harrison P, Loh M (2010). WHO guidelines for indoor air quality: Selected pollutants. Copenhagen: WHO. |
|
|
Braak H, Rüb U, Gai WP, Tredici KD (2003). Idiopathic Parkinson's disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. Journal of Neural Transmission 110(5): 517-536. |
|
|
Braak H, Tredici KD, Rüb U, Vos RA, Steur EN, Braak E (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging 24(2): 197-211. |
|
|
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu S (2004). Calcium, ATP, and ROS: A mitochondrial love-hate triangle. American Journal of Physiology-Cell Physiology 287:4. |
|
|
Catalá A, Díaz M (2016). Editorial: Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Frontiers in Physiology, Volume 7. |
|
|
Chan FK, Moriwaki K, Rosa MJ (2013). Detection of Necrosis by Release of Lactate Dehydrogenase Activity. Methods in Molecular Biology Immune Homeostasis pp. 65-70. |
|
|
Channel SR, Latendresse JR, Kidney JK, Grabau JH, Lane JW, Steel-Goodwin L, Gothaus MC (1998). A Subchronic Exposure to Trichloroethylene Causes Lipid Peroxidation and Hepatocellular Proliferation in Male B6C3F1 Mouse Liver. Toxicological Sciences 43(2):145-154. |
|
|
Chatterjee S, Chatterjee A, Roy S, Bera B, Bandyopadhyay SK (2014). L-Theanine healed NSAID-induced gastric ulcer by modulating pro/antioxidant balance in gastric ulcer margin. Journal of Natural Medicines 68(4):699-708. |
|
|
Chen S, Wang Z, Ma Y, Zhang W, Lu J, Liang Y, Zheng X (2018). Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases. Molecules 23(3):512. |
|
|
Cheng H, Ulane CM, Burke RE (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Annals of Neurology 67(6):715-725. |
|
|
Cho H, Kim S, Lee S, Park JA, Kim S, Chun HS (2008). Protective effect of the green tea component, l-theanine on environmental toxins-induced neuronal cell death. NeuroToxicology 29(4):656-662. |
|
|
Deb S, Dutta A, Phukan BC, Manivasagam T, Thenmozhi AJ, Bhattacharya P, Borah A (2019). Neuroprotective attributes of L-theanine, a bioactive amino acid of tea, and its potential role in Parkinson's disease therapeutics. Neurochemistry International 129:104478. |
|
|
DeMaagd G, Phillip A (2015). Parkinson's Disease and Its Management. Pharmacy and Therapeutics 40(8): 504-510. |
|
|
Doherty RE (2000). A History of the Production and Use of Carbon Tetrachloride, Tetrachloroethylene, Trichloroethylene and 1,1,1-Trichloroethane in the United States: Part 2--Trichloroethylene and 1,1,1-Trichloroethane. Environmental Forensics 1(2):83-93. |
|
|
Emamzadeh FN, Surguchov A (2018). Parkinson's Disease: Biomarkers, Treatment, and Risk Factors. Frontiers in Neuroscience P. 12. |
|
|
Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Prince TS (2008). Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Annals of Neurology 63(2):184-192. |
|
|
Goldman SM (2010). Trichloroethylene and Parkinson's disease: Dissolving the puzzle. Expert Review of Neurotherapeutics 10(6):835-837. |
|
|
Guehl D, Bezard E, Dovero S, Boraud T, Bioulac B, Gross C (1999). Trichloroethylene and parkinsonism: A human and experimental observation. European Journal of Neurology 6(5):609-611. |
|
|
Inamdar N, Arulmozhi D, Tandon A, Bodhankar S (2007). Parkinsons Disease: Genetics and Beyond. Current Neuropharmacology 5(2):99-113. |
|
|
Khare S, Gokulan K, Williams K, Bai S, Gilbert KM, Blossom SJ (2018). Irreversible effects of trichloroethylene on the gut microbial community and gut-associated immune responses in autoimmune-prone mice. Journal of Applied Toxicology 39(2):209-220. |
|
|
Krasnic T (2017). Preliminary Information on Manufacturing, Processing, Distribution, Use, and Disposal: Trichloroethylene [PDF]. United States Environmental Protection Agency. |
|
|
Li C, Tong H, Yan Q, Tang S, Han X, Xiao W, Tan Z (2016). L-Theanine Improves Immunity by Altering TH2/TH1 Cytokine Balance, Brain Neurotransmitters, and Expression of Phospholipase C in Rat Hearts. Medical Science Monitor 22:662-669. |
|
|
Liu M, Choi D, Hunter RL, Pandya JD, Cass WA, Sullivan PG, Bing G (2010). Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. Journal of Neurochemistry 112(3):773-783. |
|
|
Mahajan SD, Law W, Aalinkeel R, Reynolds J, Nair BB, Yong K, Schwartz SA (2012). Nanoparticle-Mediated Targeted Delivery of Antiretrovirals to the Brain. Methods in Enzymology Nanomedicine - Infectious Diseases, Immunotherapy, Diagnostics, Antifibrotics, Toxicology and Gene Medicine pp. 41-60. |
|
|
Muñoz E, Pastor P, Martí MJ, Valldeoriola F, Tolosa E, Oliva R (2001). Enfermedad de Parkinson esporádica y familiar: Estudio comparativo. Medicina Clínica 116(16):601-604. |
|
|
Nango H, Kosuge Y, Miyagishi H, Sugawa K, Ito Y, Ishige K (2017). Prostaglandin E2 facilitates neurite outgrowth in a motor neuron-like cell line, NSC-34. Journal of Pharmacological Sciences 135(2):64-71. |
|
|
Rietdijk CD, Perez-Pardo P, Garssen J, Wezel RJ, Kraneveld AD (2017). Exploring Braak's Hypothesis of Parkinson's Disease. Frontiers in Neurology P. 8. |
|
|
Russell HH, Matthews JE, Sewell GW (1992). TCE Removal from Contaminated Soil and Ground Water [PDF]. United States Environmental Protection Agency. |
|
|
Saqcena M, Menon D, Patel D, Mukhopadhyay S, Chow V, Foster DA (2013). Amino Acids and mTOR Mediate Distinct Metabolic Checkpoints in Mammalian G1 Cell Cycle. PLoS ONE 8:8. |
|
|
Sharma N, Nehru B (2017). Curcumin affords neuroprotection and inhibits α-synuclein aggregation in lipopolysaccharide-induced Parkinson's disease model. Inflammopharmacology 26(2):349-360. |
|
|
Sharma RK, Candelario-Jalil E, Feineis D, Bringmann G, Fiebich BL, Akundi RS (2017). 1-Trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo) Alters Cell Cycle Progression in Human Neuroblastoma Cell Lines. Neurotoxicity Research 32(4):649-660. |
|
|
Sugiyama T, Sadzuka Y, Tanaka K, Sonobe T (2001). Inhibition of glutamate transporter by theanine enhances the therapeutic efficacy of doxorubicin. Toxicology Letters 121(2):89-96. |
|
|
Thangarajan S, Deivasigamani A, Natarajan SS, Krishnan P, Mohanan SK (2014). Neuroprotective activity of L-theanine on 3-nitropropionic acid-induced neurotoxicity in rat striatum. International Journal of Neuroscience 124(9):673-684. |
|
|
Triarhou LC (2013). Dopamine and Parkinson's Disease. Austin, Texas: Landes Bioscience. |
|
|
Yokogoshi H, Terashima T (2000). Effect of theanine, r-glutamylethylamide, on brain monoamines, striatal dopamine release and some kinds of behavior in rats. Nutrition 16(9):776-777. |
|
|
Yulug B, Ozansoy M, Cankaya S (2019). A different view on the pathophysiology of Parkinson's disease: A descendent neurochemical hypothesis? Neural Regeneration Research 14(10):1717. |
|
|
Zaheer F, Slevin JT (2011). Trichloroethylene and Parkinson Disease. Neurologic Clinics 29(3):657-665. |
|
|
Zhu Q, Shen T, Ding R, Liang Z, Zhang X (2005). Cytotoxicity of trichloroethylene and perchloroethylene on normal human epidermal keratinocytes and protective role of Vitamin E. Toxicology 209(1):55-67. |
|
|
Ziv I, Zilkha-Falb R, Offen D, Shirvan A, Barzilai A, Melamed E (1997). Levodopa induces apoptosis in cultured neuronal cells-A possible accelerator of nigrostriatal degeneration in Parkinson's disease? Movement Disorders 12(1):17-23. |
|
Copyright © 2024 Author(s) retain the copyright of this article.
This article is published under the terms of the Creative Commons Attribution License 4.0


