Aldrin used in agricultural as well as industrial or home pest control is known to be a persistent organic pollutant, having chronic adverse effects on wildlife and humans. The degradation of aldrin has drawn a lot of attentions. In this study, house fly cytochrome P450 6A1 (CYP 6A1) or both house fly cytochrome P450 6A1 and human NADPH-P450 reductase (CPR) together was expressed in Escherichia coli cells to study their activity on aldrin conversion. It was found out that the membrane fractions prepared from the recombinant E. coli cells expressing human CPR with CYP 6A1 have catalytic activity on aldrin using enzyme analysis. The catalytic activity of the recombinant E. coli cells was also investigated on aldrin. After adding aldrin to the co-expression of human CPR and CYP 6A1 recombinant E. coli cells culture with 1 µmol L-1 final concentration, 163.36 nmol L-1 of dieldrin was detected after 8 h, the product of aldrin epoxidation. These results indicate that co-expressing house fly CYP 6A1 and human CPR can convert aldrin to dieldrin.
Aldrin is an organochlorine insecticide which has been used in some developing countries to kill rootworms, beetles, and termites because of its low cost and ability in controlling insects (Costa, 2015). The difficulty to degrade aldrin leads to its extremely accumulation in environment and living organisms (Jhamtania et al., 2018). Thus, aldrin pollution still remains a serious problem in environment and it is required to find some efficient methods for its remediation. Degrading aldrin by microorganisms including bacteria and soil fungi has been reported (Erick et al., 2006; Ferguson et al., 1977; Xiao et al., 2011). The possibility of using Pseudomonas fluorescens cell cultures to biodegrade aldrin in water, obtaining 94.8% degradation of aldrin in 120 h was demonstrated (Erick et al. 2006). Ferguson et al. (1977) reported that members of the order Actinomycetales can epoxidize aldrin to exo-dieldrin with their bacterial enzyme system (Ferguson et al. 1977). Xiao et al. (2011) investigated Phlebia acanthocystis, Phlebia brevispora, and Phlebia aurea to value their degradation capacities on aldrin and the subsequent metabolic products. Over 90% of aldrin was degraded after 28 days and several new metabolites including 9-hydroxyaldrin and two carboxylic acid products were also detected in the fungal cultures (Xiao et al., 2011).
Although using natural microorganisms to degrade aldrin in polluted soil is promising, there has been no report of the use of engineered microorganisms to degrade aldrin till now. The construction of an aldrin-metabolizing enzyme and its over-expression in microorganisms are useful for bioremediation. Cytochrome P450s from insects are involved in the metabolism of synthetic insecticides (Goff et al., 2003), secondary plant chemicals (Wiseman and Lewis, 2007), and insect hormones (Liu et al., 2015). The characteristics of insect P450s indicate that they can be used for the study of the metabolism and biodegradation of insecticides. It has been reported that phenobarbital treatment increases cytochrome P450 6A1 gene levels in house fly and increases the epoxidation of cyclodiene insecticides in particular (
Cariño et al., 1994). This suggests that cytochrome P450 6A1 are related with aldrin metabolism. A homogeneous recombinant cytochrome P450 6A1 from house fly was obtained by expressing it in
Escherichia coli and was exploited to monitor aldrin pollution in the environment (Wu et al., 2011; Zhang et al., 2010). However, microsomal P450s depend on their redox partner, NADPH-cytochrome P450 reductase (CPR), to deliver electrons from nicotinamide adenine dinucleotide phosphate (NADPH). In eukaryotic cells, the endogenous electron transport system is enough to support the P450’s activity (Zhang et al., 2017); however, the reduction rate of heme iron of cytochrome P450s is very low because of the insufficient endogenous electron transport system in
E. coli. To enhance the
in vivo catalytic activity of P450 in
E. coli, addition of CPR is required. It was reported that the co-expression of cytochrome P450 and cytochrome-450 reductase in the same
E. coli cells can eliminate the need to add reductase (Grinkova et al., 2010). Like cytochrome P450s, cytochrome-450 reductase can be easily expressed in
E. coli with certain activity (Kimura and Iyanagi 2003). Considering the availability of human cytochrome P450 reductase gene in our laboratory, choice was made to express house fly cytochrome P450 6A1 and human cytochrome P450 reductase in
E. coli in the present study. It was found out that the genetically engineered
E. coli strains have metabolic activity of converting aldrin to dieldrin by gas chromatography-mass spectrometry (GC-MS) analysis.
The expression vector pCW and human cpr/pCW were given by Dr. F. Peter Guengerich (Medical Center of Vanderbilt University). The gene cyp6a1 and E. coli DH5α cell were preserved in our laboratory. Aldrin, lysozyme, dithiothreitol (DTT) and Triton N-101 were bought from Sigma, America. δ-Aminolevulinic acid (δ-ALA) was obtained from Aldrich, Merck. The restriction enzymes, NdeI and HindIII, and Isopropyl-β-D-thiogalactopyranoside (IPTG) were from Takara, Japan. Terrific broth (TB) and Luria-Bertani (LB) media were from Invitrogen, America. All other chemicals were of reagent grade and were used directly without other treatment.
Construction of co-expression plasmids for house fly CYP6A1 and human CPR
The cyp6a1/pCW was constructed and preserved in our laboratory. Full length of cyp6a1 was cloned from house fly, digested with NdeI/SalI and then ligated into NdeI/SalI-cut the vector pCW (Zhang et al., 2010). In this study, NdeI and HindIII sites were created for cyp6a1 by PCR. The forward 5′- AGCGACATATGGCTTTTGGTTCATTTCT-3′ (the underlined sequence is the NdeI site) and reverse 5′- GCCAAGCTTTTATTTAATTTTCTTTCTT -3′ (the underlined sequence is the HindIII site) were used as the primers. A full-length human cpr was obtained by digesting with HindIII enzyme from cpr/pCW vector. To link cyp6a1 with cpr, the 16 bp sequence TAACTTTAAGAAGGAGAT, which contained an optimized Shine-Dalgarno ribosomal binding site (RBS), was designed to enhance its production in E. coli.
Expression of house fly CYP6A1 and human CPR
A single ampicillin-resistant colony of E. coli BL 21 cells transformed with plasmid DNA was grown overnight at 37°C in Luria-Bertani medium supplemented with 100 ug mL-1 ampicillin. A 1 mL aliquot was used to inoculate each 1.0 L of the TB containing 0.02% bactopeptone (w/v). The TB medium was supplemented with ampicillin (50 ug mL-1), 1.0 mM thiamine, 0.5 mM δ-ALA and trace elements [27 g FeCl3·6H2O, 2.0 g CoCl2·6H2O, 2.0 g ZnCl2·4H2O, 2.0 g Na2MoO4, 1.0 g CaCl2·2H2O, 1.0 g CuCl2, 0.5 g H3BO3, and 100 mL concentrated HCl in 1 L ultrapure water]. Induction of the tac promoters was done by adding 1.0 mM IPTG when the OD600 of the expression culture reached 0.6. Then it was shaken at 37°C and 180 rpm and the culture was incubated for another 36 h at 28°C and 160 rpm in a shaker.
Cell harvesting and lysis
The pelleted cells were resuspended in 2×TSE (100 mM Tris-acetate, pH 7.6, containing 500 mM sucrose, and 0.5 mM EDTA) according to 5 mL of 2×TSE versus 100 mL of culture. Following the addition of lysozyme (0.25 mg/mL), the cells were gently shaken for 30 min after the addition of an equal volume of H2O. The lysed cells were centrifuged by 2800 g, 4°C for 15 min. The resulting spheroplasts (4 mL/100 mL of culture, in 100 mM pH 7.4 potassium phosphate buffer), which contains 6 mM magnesium acetate, 20% glycerol (v/v) and 0.01 mM DTT, were frozen at -70°C until further use. During the thawing phase, the protease inhibitors of PMSF were added at final concentration of 2 μΜ. Cells were sonicated for 20 min with 5 s bursts (at 50% full power) while they were kept in an ice-salt bath. The resulting lyaste was centrifuged at 12000 rpm for 12 min, and the resulting supernatant was then centrifuged at 180000 g for 65 min; the pellet (that is, membranes), which contains 0.25 mM EDTA and 0.25 M sucrose, was resuspended in 20 mM Tris-acetate buffer (pH 7.4) and then was stored at -70°C.
Characterization of CYP6A1 and CPR expression
CYP 6A1 content was calculated by the general Omura and Sato method using an extinction coefficient of 91 mM-1 cm-1 (Omura, 1964). Total protein concentration was determined by the Lowry assay (Lowry et al., 1951). Activities of CPR were determined by the reduction of cytochrome c (20 µM) with addition of 100 µM NADPH. All assays and incubations were carried out in a 100 mM pH 7.4 potassium phosphate buffer. The reduction rate of CPR was monitored by the increased absorbance at 550 nm (Guengerich et al., 2009). For calculating the reduction rate of CPR, a specific molar absorption coefficient (21.1 mM-1 cm-1) of equine heart cytochrome c was used.
Measurement of catalytic activity of CYP6A1 and CPR towards aldrin
The reaction mixture contained 2 to 200 µM aldrin (dissolved in acetone) and 50 nM of CYP6A1 and 7 nM of CPR in a final volume of 1.0 mL 50 mM pH 7.4 potassium phosphate buffer. The reaction was initiated after adding NADPH with 0.5 mM final concentration. Incubations were carried out at 37°C for 30 min and terminated by vortexing with 4 mL of ethyl acetate, and 5 µM heptachlor was added to the mixture as an internal standard for GC-MS analysis. Aliquots of the reaction mixture were collected at varying time intervals up to 30 min and the reaction product was extracted with four volumes of petroleum ether. The organic phase was recovered and dried up by a rotary evaporator. The resulting residue was resolubilized with 50 µL of petroleum ether. A 1 µL aliquot was injected into GC-MS equipped with a DB-5MS capillary column and an electron capture detector. Helium was used as the carrier gas with a flow of 36.8 cm s-1. Samples were injected automatically in splitless mode by HP 7673 auto sampler with 1.00 µL each time. The injection port temperature was kept at 260°C. The column temperature was maintained at 80°C for 1 min, and then programmed at 25°C min-1 to 200°C, which was held for 1 min, then at 5°C min-1 to 250°C, and then increase at 20°C min-1 to 290°C, which was held for 5 min. The mass-spectrometer acquisition parameters were as follows: electron energy 70 eV, solvent delay 10 min, electron multiplier voltage 2000 V. Metabolism product was detected with selected ion monitoring (SIM).
Determination of catalytic activity of the recombinant E. coli cells towards aldrin
The substrate aldrin was dissolved in acetone. To study the metabolism of aldrin by the recombinant E. coli cells, the final concentration was adjusted to 1 µM. The recombinant E. coli cells were cultivated at 30°C after adding aldrin. At varying time intervals, up to 8 h aliquots of the cell culture were sampled and extracted with four volumes of diethyl ether-petroleum ether (1:1). The organic phase was collected then dried up. The resulting residue was resolubilized with petroleum ether and applied to the aforementioned GC-MS analysis. Samples were analyzed three times in the same assay and in three different runs. The mean data was obtained to analyze the catalytic activity of the recombinant E. coli cells towards aldrin.
Construction and expression of CYP6A1 and CPR in E. coli
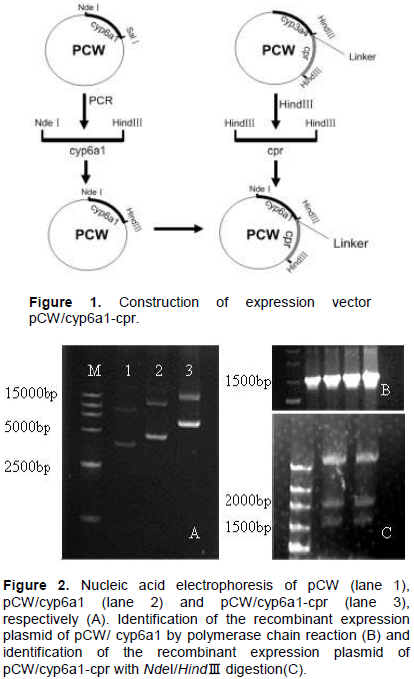
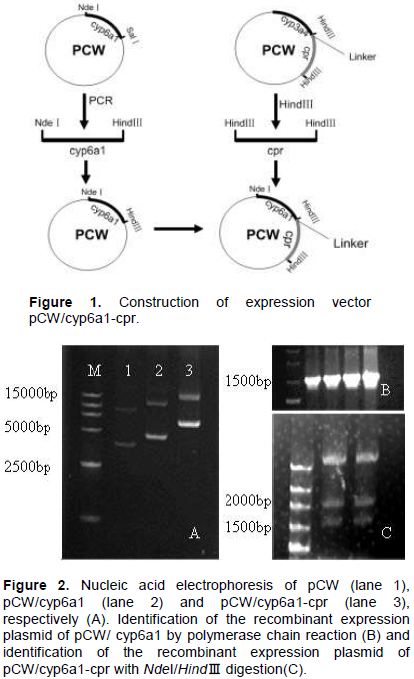
Figure 1 shows the schemes to construct the expression plasmid. cyp6a1 and cpr gene fragment was cut from the recombinant plasmid pCW with NdeI/HindIII and HindIII, respectively and subcloned into the expression vector pCW. A 16 bp linker containing a Shine-Dalgarno ribosomal binding site was employed to link cyp6a1 and cpr. After ligation and transformation, the positive recombinants were subsequently screened out by using ampicillin selective media.Figure 2A shows the nucleic acid electrophoresis of pCW, pCW/CYP6A1 and pCW/CYP6A1-CPR, respectively. The CYP6A1 recombinants were identified by PCR with the forward and reverse primers (Figure 2B). The CYP6A1 and CPR recombinants were proven to contain the 1.5 and 2.0 kb fragments by enzyme cutting with NdeI and HindIII, respectively (Figure 2C).
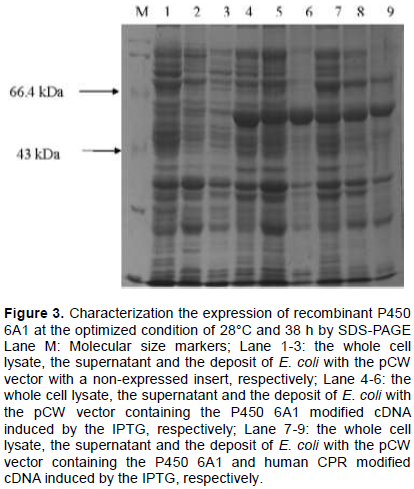
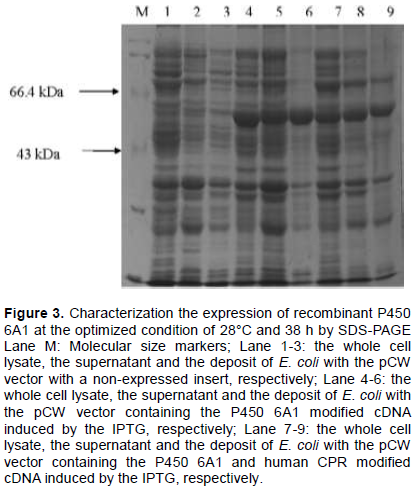
The temperature and time for E. coli growth were optimized and the highest expression level was obtained at 28°C and 38 h. Compared with no insertion, the expression of CYP 6A1 was obvious (Figure 3). The molecular weight of P450 6A1 is about 58 kDa, which is consistent with the gene product. However, the expression of CPR is too low to be detected by SDS-PAGE.
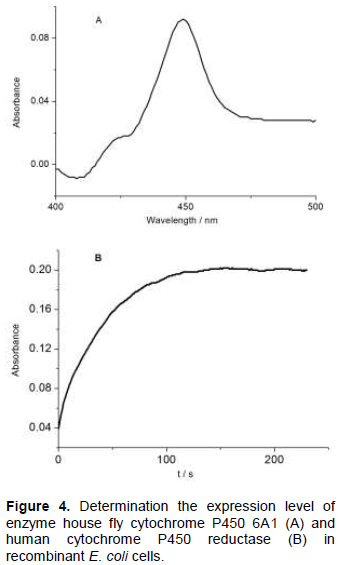
The determination of the CYP6A1 content was based on the method of Omura and Sato (1964) and the result is shown in Table 1. As shown in Figure 4A, the membrane fraction of CYP6A1 showed a maximum absorption peak at 447 nm with a small peak at 420 nm.
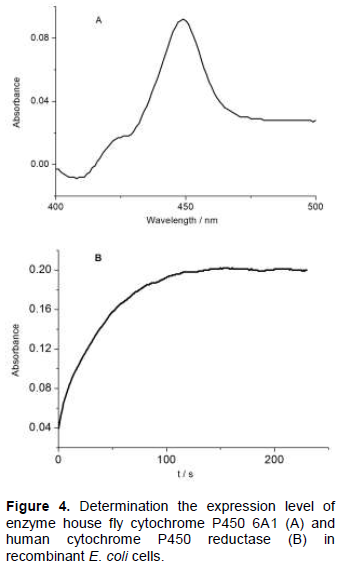
The results indicate the recombinant protein was produced in good quality. On the basis of reduced CO difference spectrum, the expression level in the membrane fraction of CYP6A1 in cyp6a1/pCW construct or in cyp6a1-cpr/pCW construct was estimated to be 350 nmol L-1 or 158 nmol /L-1 culture, respectively. The expression level of P450 in CYP6A1-CPR co-expression is almost half of that in the expression of P450 alone. The limit of the capacity of protein synthesis in E. coli for the additional two proteins synthesis may cause the decrease in the expression level of P450. Compared with the previous reported data about the expression level of cytochrome P450s (Hiroshi et al., 1998), the highest expression level of CYP2C8 with 381 nmol L-1 of culture and the lowest expression level of CYP2A6 with 66 nmol L-1 of culture, the expression level of CYP6A1 in this report is medium.
Cytochrome c reductase activity was used to characterize the expression level of CPR in the recombinant E. coli cells (Figure 4B). From the results in Table 1, it can be concluded that the cytochrome c reductase activity in the recombinant E. coli cells expressing both CYP6A1 and CPR was 137-fold higher than that expressing CYP6A1 only. In the previous report, the expression level of human CPR, co-expressed with cytochrome P450, ranged from 68 to 312 nmol L-1 of culture (Hiroshi et al., 1998). The expression level of CPR in this report is very low. The low yield of CPR in this report may be caused by the case of co-expression of proteins.
Metabolism of aldrin by CYP6A1 and CYP6A1/CPR
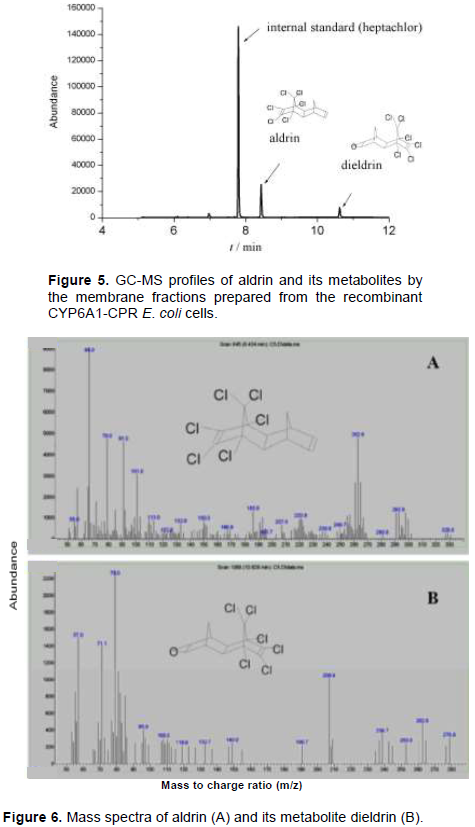
CYP6A1 or CYP6A1-CPR in membranes collected from the recombinant E. coli cells were examined on the metabolism of aldrin. GC-MS method was applied to analyze the metabolites as reported previously (Xiao et al., 2011). The membrane fraction prepared from the control pCW and cyp6a1/pCW showed no activity on aldrin in this study. Figure 5 shows GC-MS profiles of the metabolites by the reconstituted CYP6A1/CPR. At 30 min of incubation, 25% of aldrin was converted into its metabolite. Mass spectrum of aldrin (retention time 8.4 min) and its metabolite (retention time 10.6 min) is as shown in Figure 6. Aldrin with molecular weight 365 has a mass spectrum showing the following fragment ion results: m/z 329[M-Cl]+, m/z 293 [329-HCl]+, and 263 [293-CH2CH3]+. Its metabolite had fragment ions at m/z 345 [M-HCl]+, m/z 263 [345-OCH2CH3Cl]+, m/z 237 [C5Cl5]+, and 79. Based on comparison with published data (Xiao et al., 2011), it was concluded that metabolite was dieldrin, the epoxidized product of aldrin.
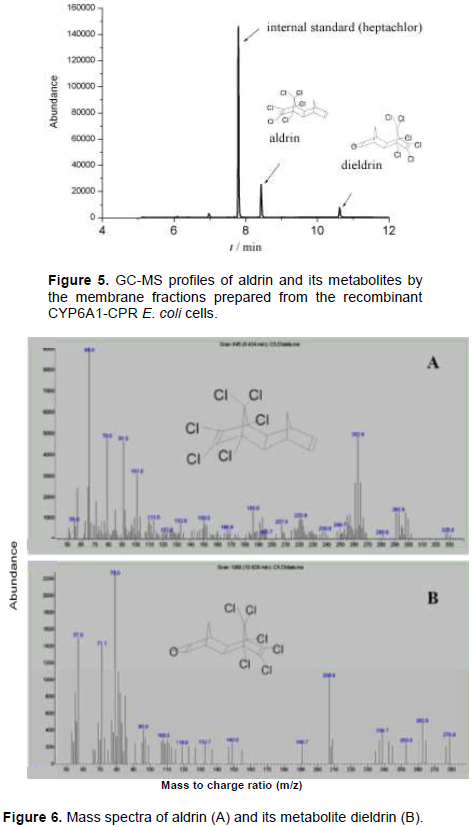
According to the product dieldrin turnover and the double reciprocal method, the kinetic parameters, Km and Vmax, for the CYP6A1/CPR dependent epoxidation activity towards aldrin were estimated to be 46.1 μM and 27.3 nmol/nmol P450/min, respectively. The kinetic study using membrane fractions demonstrated that the enzymatic affinity of cytochrome P450 6A1 was higher than that of CYP6A1 immobilized in dioctadecyl dimethyl ammonium bromide (DDAB) film (Wu, 2011). However, aldrin epoxidase from rat liver was also examined on the activity of epoxidized aldrin with a Vmax of 5.11 nmol/nmol P450/min and a Km of 1.64 μM (Norie et al., 1984). The results indicate that the affinity of aldrin epoxidase with aldrin from rat liver microsomes is much higher than that of CYP6A1.
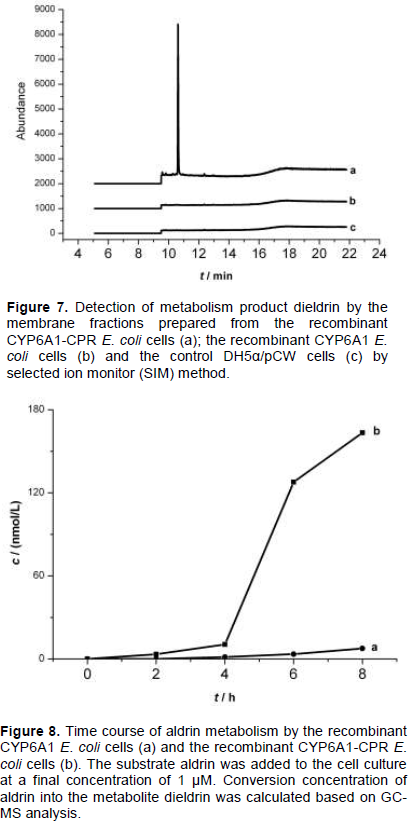
The study of the metabolism of aldrin by membrane fractions prepared from the recombinant E. coli cells indicated that CYP 6A1 alone has no activity to aldrin, but expression of human CPR with CYP 6A1 in the presence of NADPH has enzymatic activity to aldrin (Figure 7). The results indicate that human CPR played an important role in the CYP 6A1 enzyme activity.
Metabolism of aldrin by the recombinant E. coli cells
The control DH5α/pCW cells and the recombinant E. coli cells expressing CYP6A1 or CYP6A1/CPR were examined for the metabolism of aldrin. 1 µM aldrin was added to the cells culture after the cells were in logarithmic growth period. The addition of aldrin caused no growth inhibition in both recombinant cells and the control BL21/pCW cells. Cell culture (2 mL) was sampled every 2 h, then it was processed for GC-MS analysis described earlier. As shown in Figure 8, remarkable metabolism was observed towards aldrin in the recombinant CYP6A1/CPR E. coli cells, and lower activity was observed in the recombinant CYP6A1 E. coli cells, while no activity was observed in the control BL21/pCW cells. When aldrin was added into the recombinant CYP6A1/CPR E. coli cells, the epoxidation production of aldrin, dieldrin can be detected after 2 h. It indicated that the hydrophobic substrate and product can diffuse into and out of the membranes of E. coli cells. Expression of CYP6A1 alone in E. coli showed low catalytic activities in the oxidation of aldrin, indicating that some similar factors like reductase existed in E. coli cells with the ability to deliver electrons to P450. However, the co-expression of CPR and CYP 6A1 in E. coli cells led to a big increase in the catalytic activity of P450. It indicates that E. coli strains co-expressing house fly CYP 6A1 and human CPR can efficiently catalyze aldrin without addition of CPR in culture. From Figure 8, it was observed that approximately 15% of aldrin was degraded within 8 h. The results suggest that the human CPR can efficiently give electron to house fly CYP 6A1 in E. coli inner membranes to maintain the highest activity of house fly CYP 6A1.
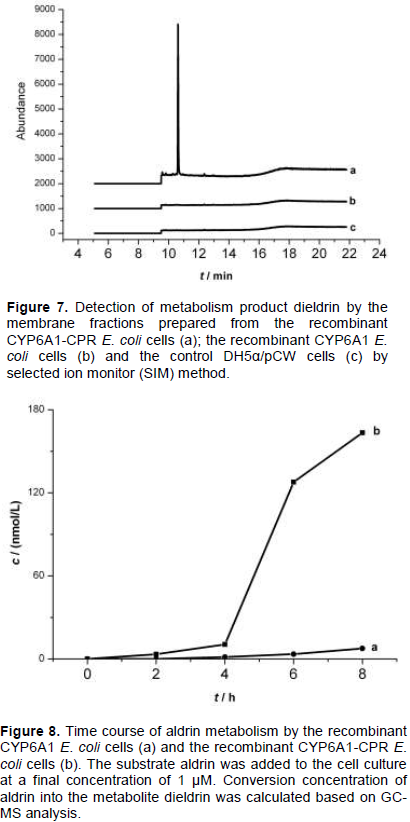
In this study, cytochrome P450 6A1 was expressed from house fly or both house fly cytochrome P450 6A1 and human cytochromes reductase in E. coli cells. It was demonstrated that the recombinant E. coli membrane fractions containing CYP6A1 have no catalytic towards aldrin, but the recombinant E. coli membrane fractions containing CYP6A1-CPR can metabolize aldrin to dieldrin. These results indicate that CPR plays an important role in the catalytic reaction of CYP6A1. The control DH5α/pCW cells and the recombinant E. coli cells expressing CYP6A1 or CYP6A1/CPR on the metabolism of aldrin have also been examined. The results show the recombinant CYP6A1/CPR E. coli cells can metabolism towards aldrin remarkably and the recombinant CYP6A1 E. coli cells show lower activity, while the control BL21/ pCW cells show no activity. These results in this study show the promise of using engineered microorganisms to degrade aldrin by expressing both CYP6A1 and CPR.
The authors have not declared any conflict of interests.
The authors are grateful for the financial support from the National Natural Science Foundation of China (No. 31670372) and the Special Fund for Basic Scientific Research of Central Colleges, South-Central University for Nationalities (CZZ18004).